
Pseudo-Reference-Based Assembly of Vertebrate Transcriptomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset and Preprocessing
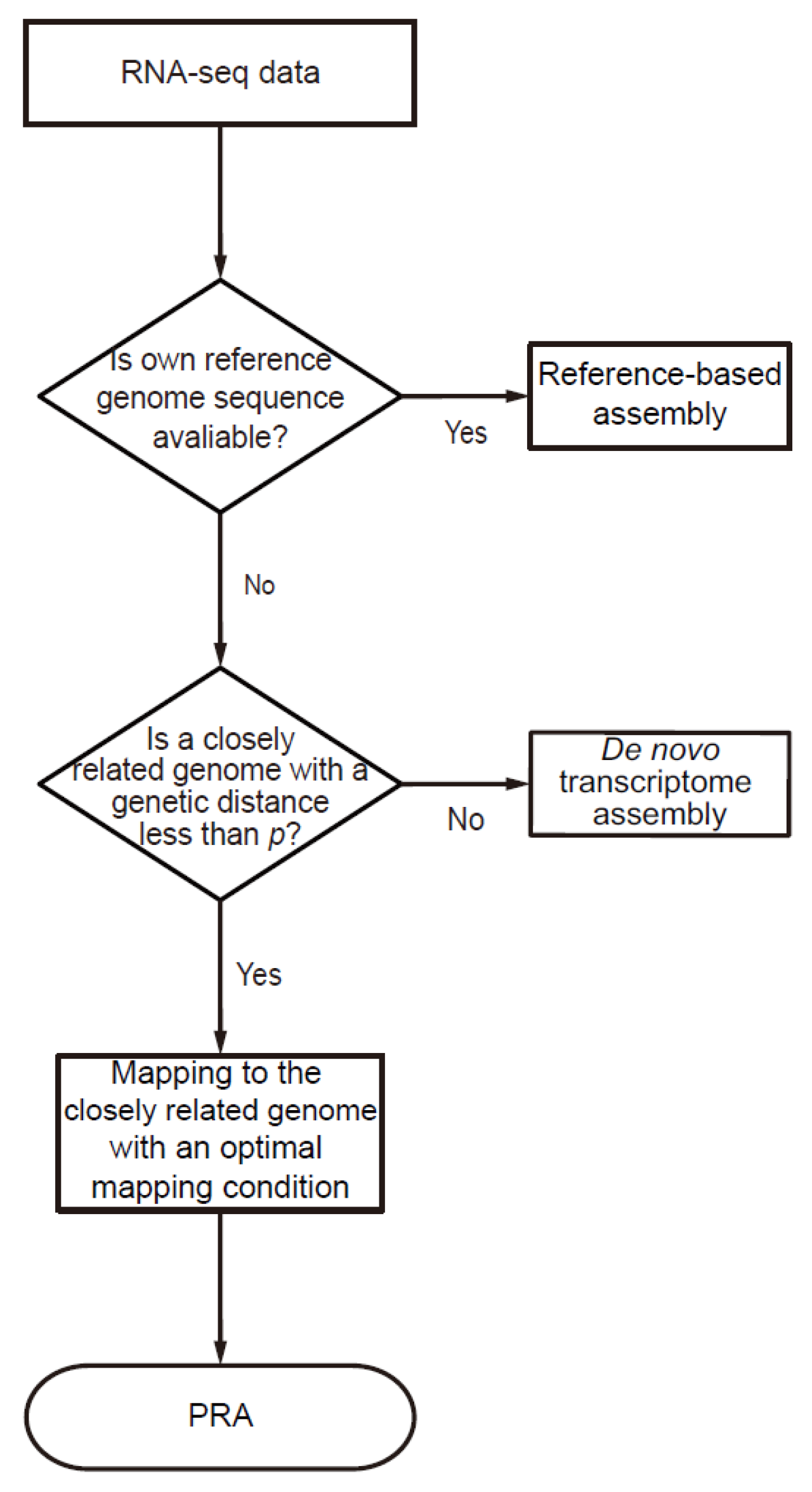
2.2. Decision Flowchart for Pseudo-Reference-Based Assembly
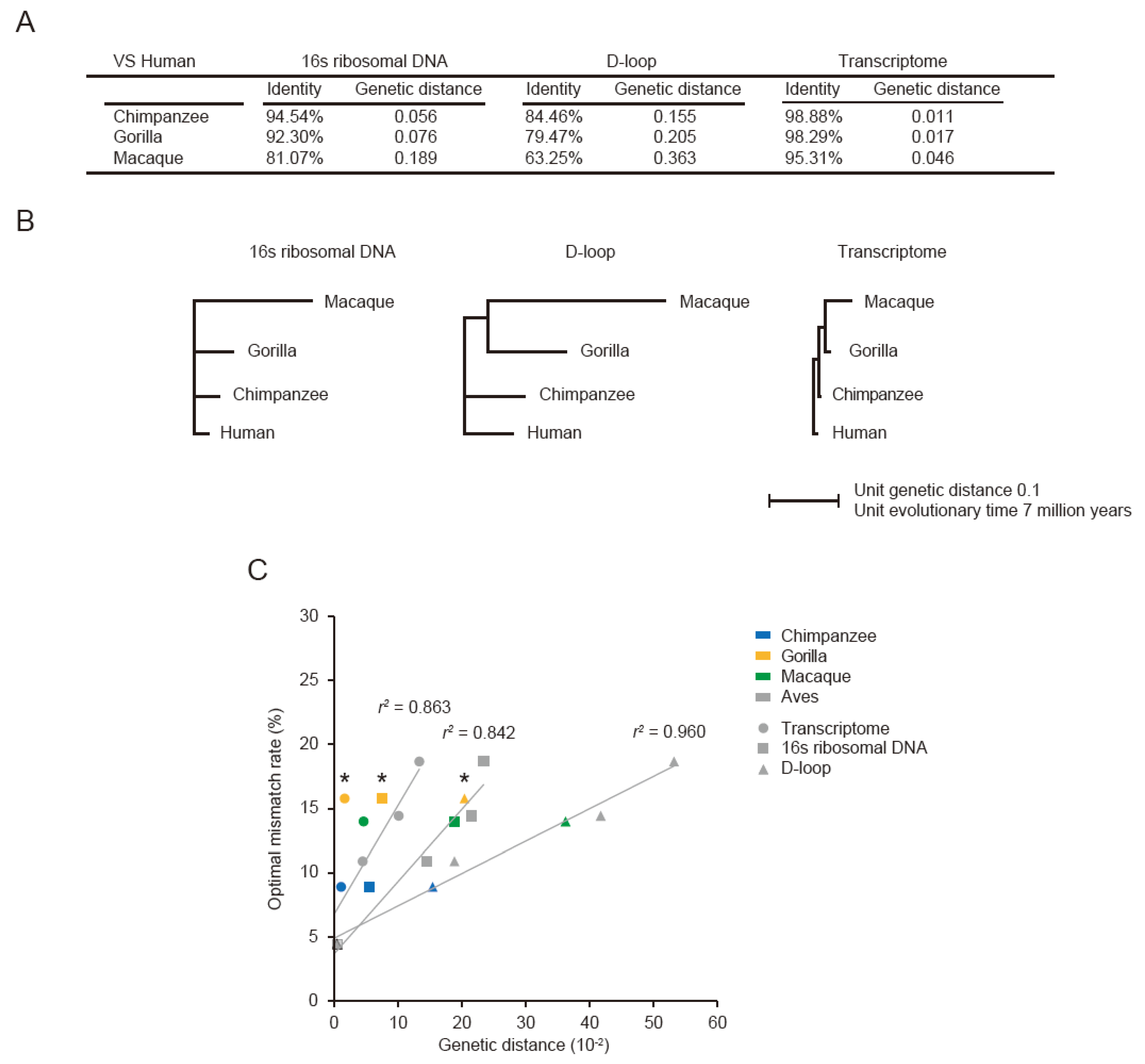
2.3. Phylogenetic Analysis of Evolutionary Marker Sequences
2.4. Transcriptome Assembly
2.4.1. Reference-Based Transcriptome Assembly and PRA
2.4.2. De Novo Transcriptome Assembly
2.5. Virtual Chicken Genome with Random Substitution
2.6. Quality Evaluation of Transcriptome Assembly
3. Results
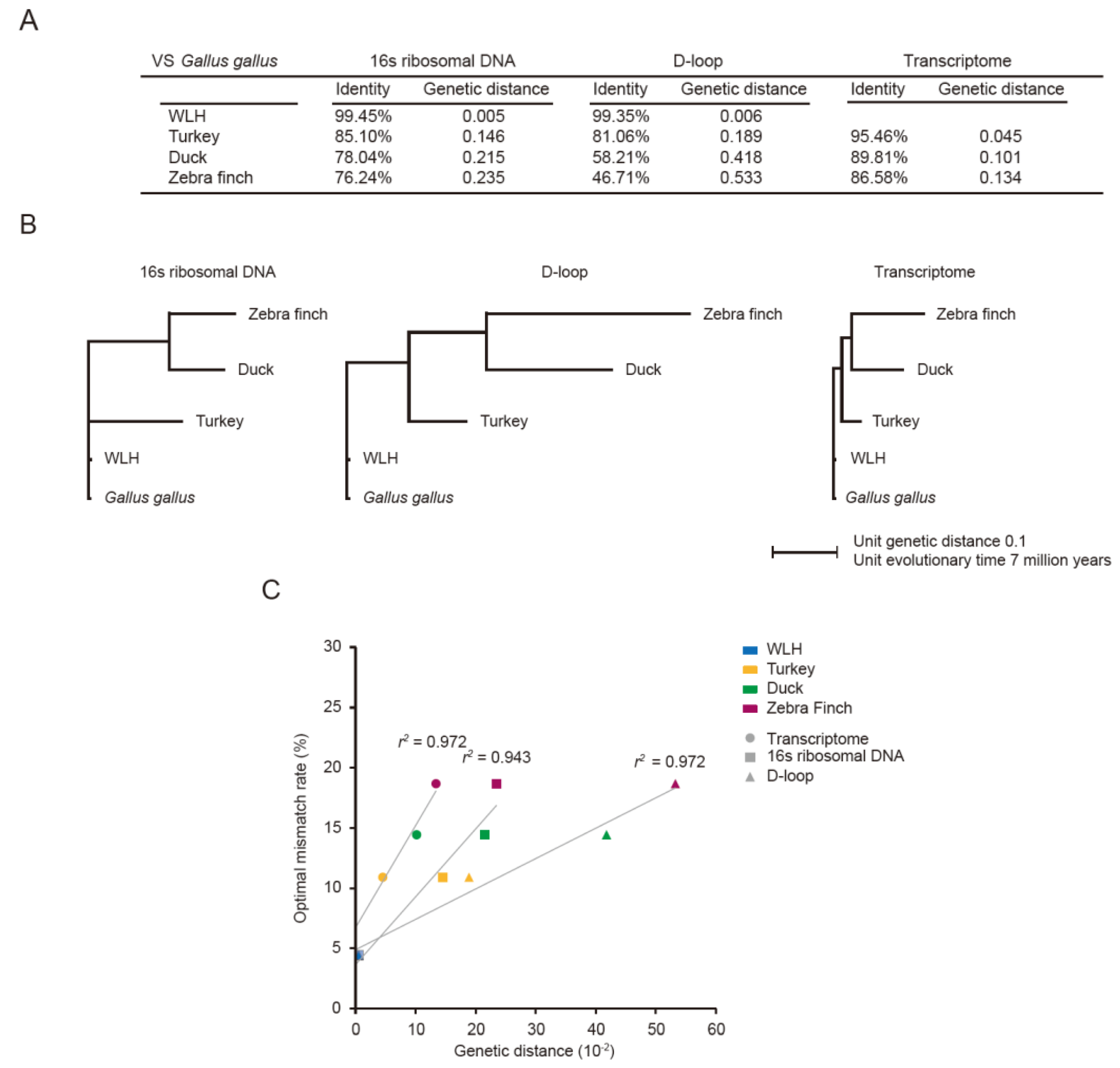
3.1. Optimal Mismatch Rates for Aves PRA
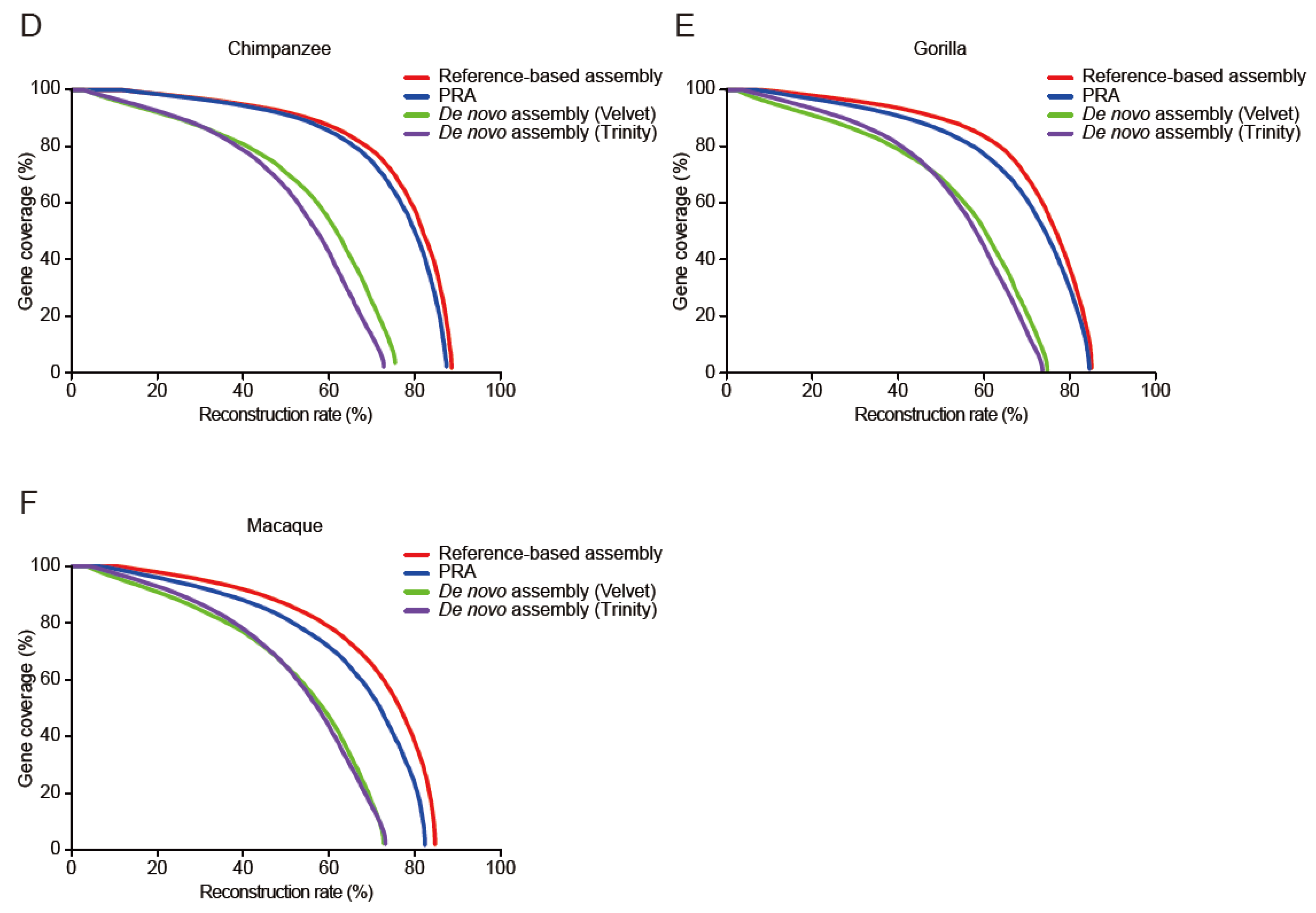
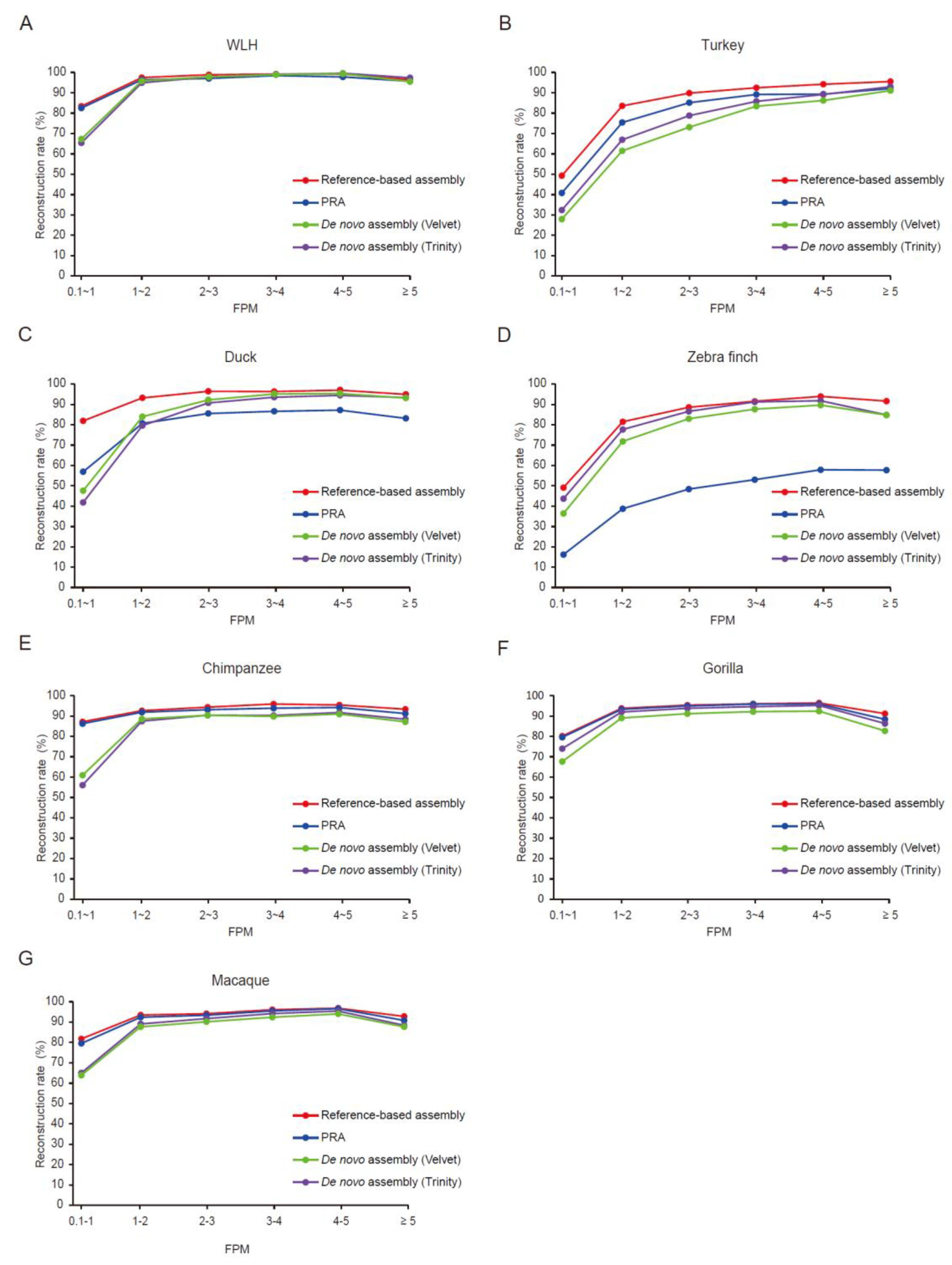
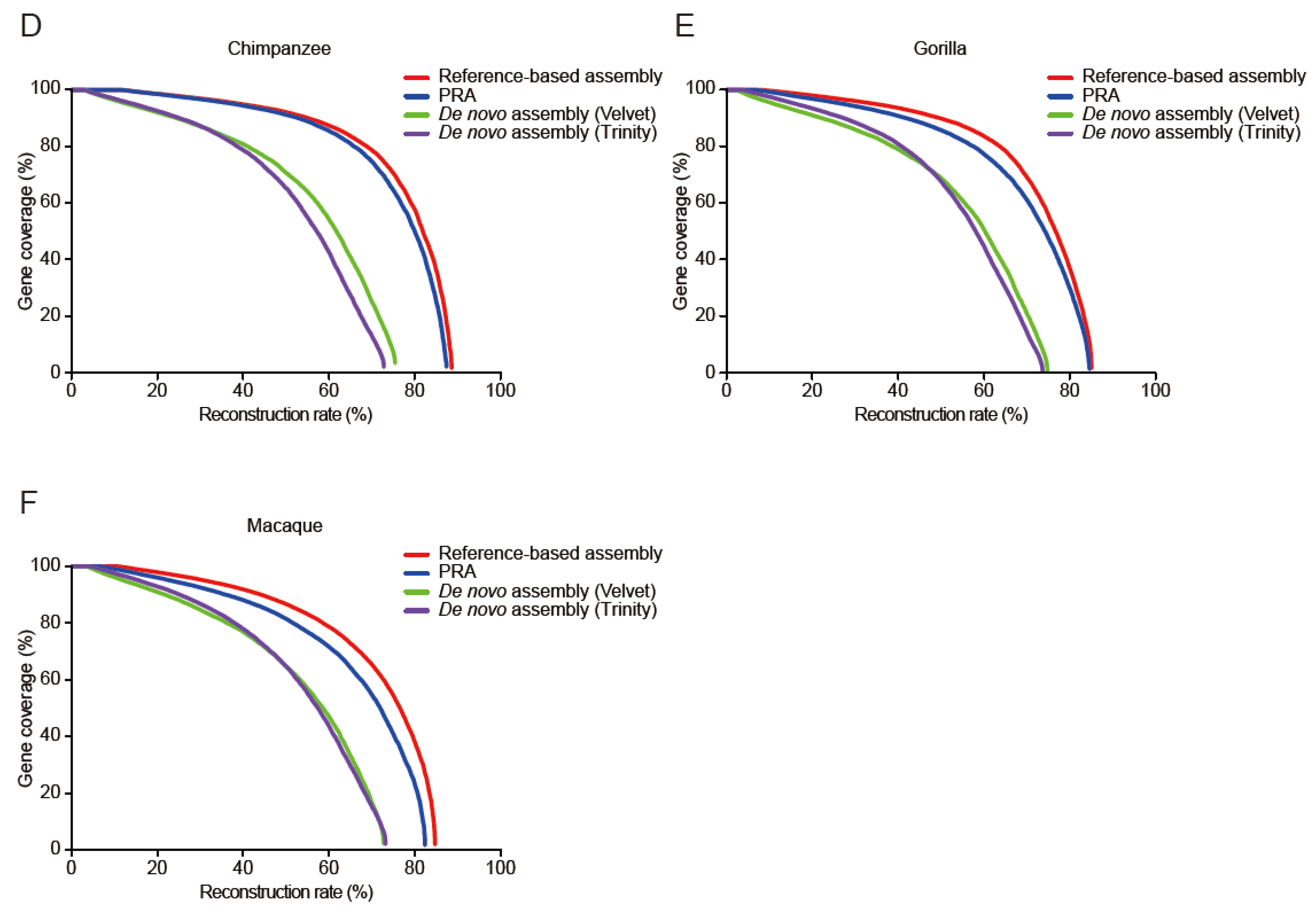
3.2. Evaluation of PRAs
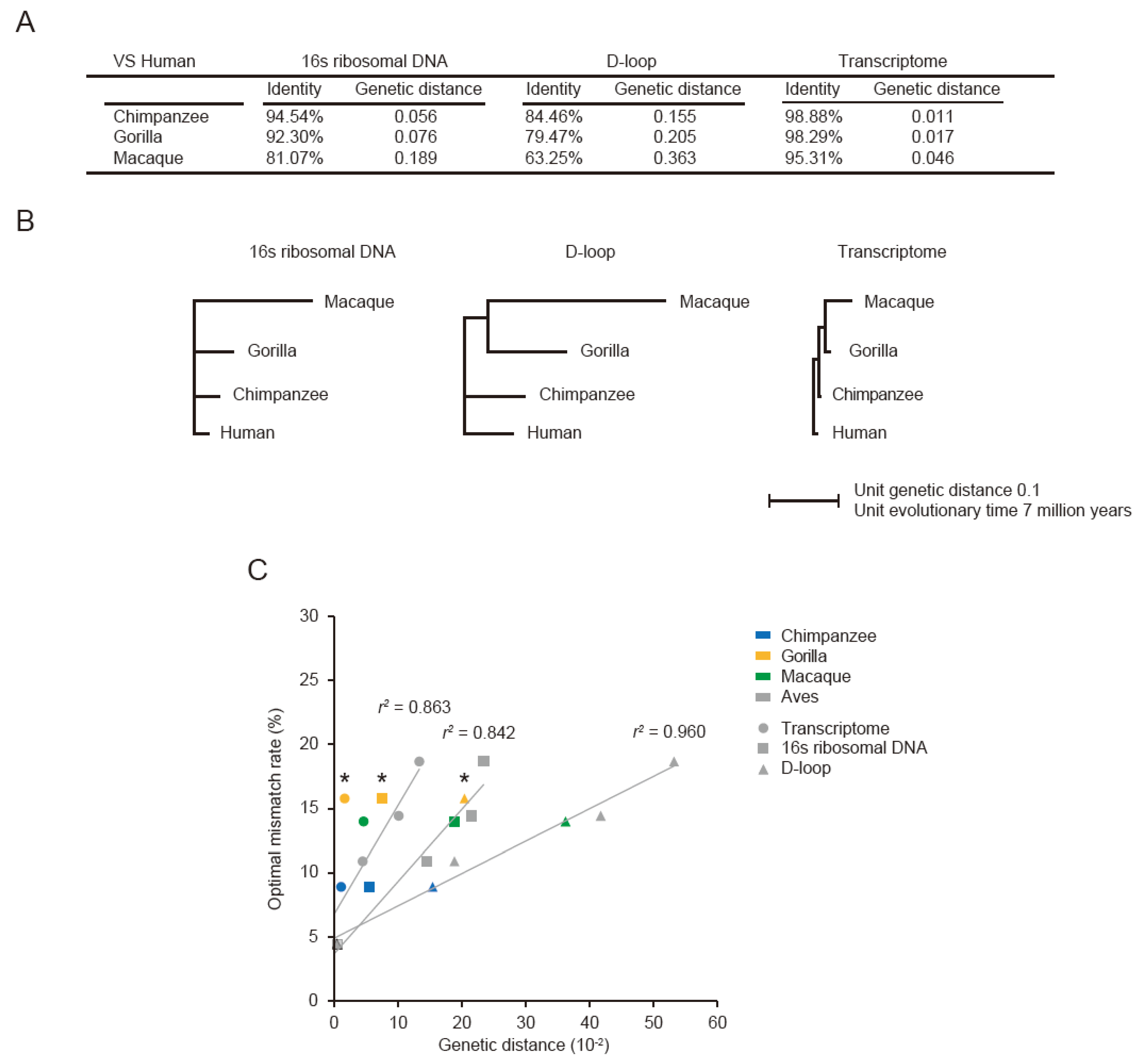
3.3. Optimal Mismatch Rates for Vertebrate PRAs
3.4. Expression Level Affects the Quality of Assembled Transcripts
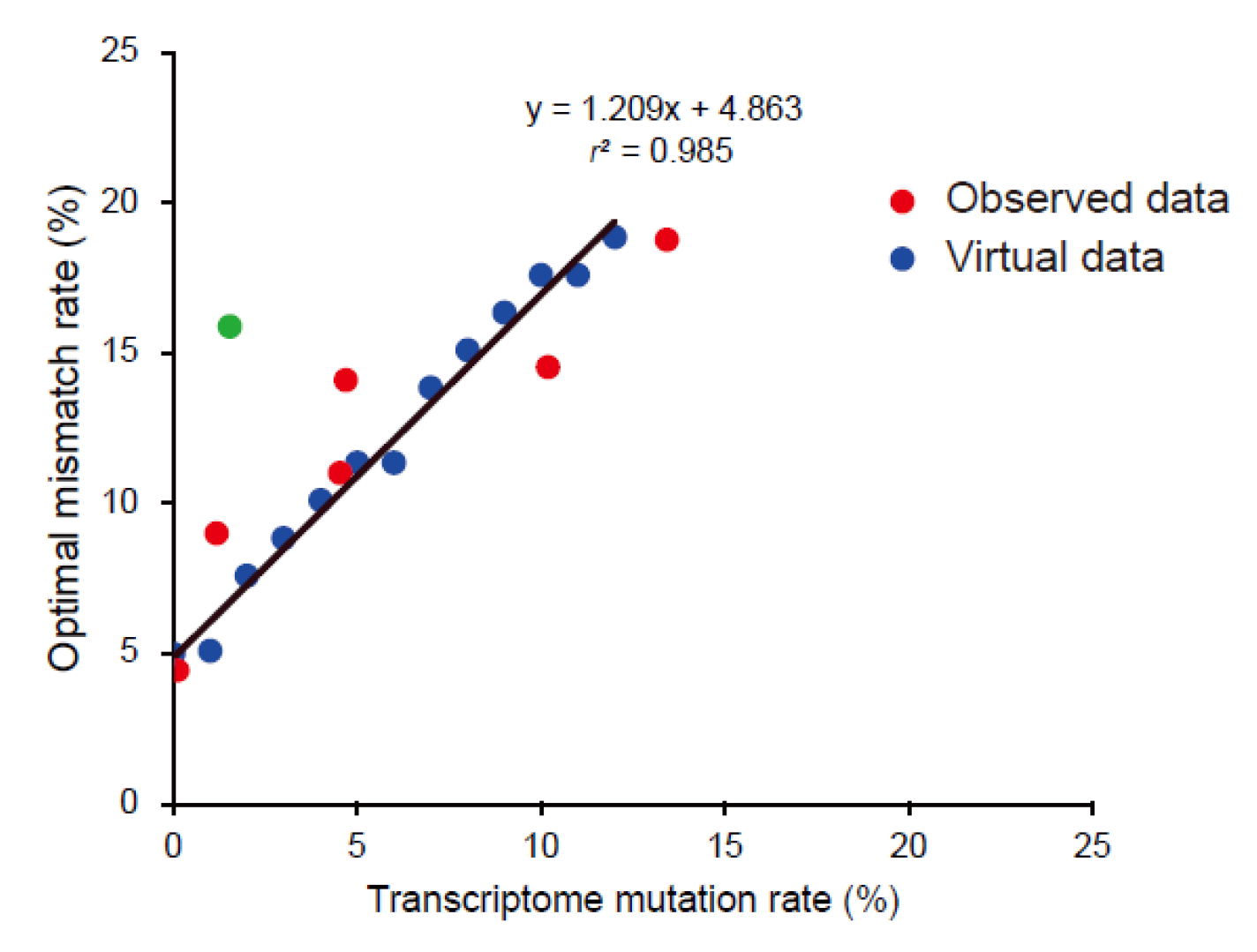
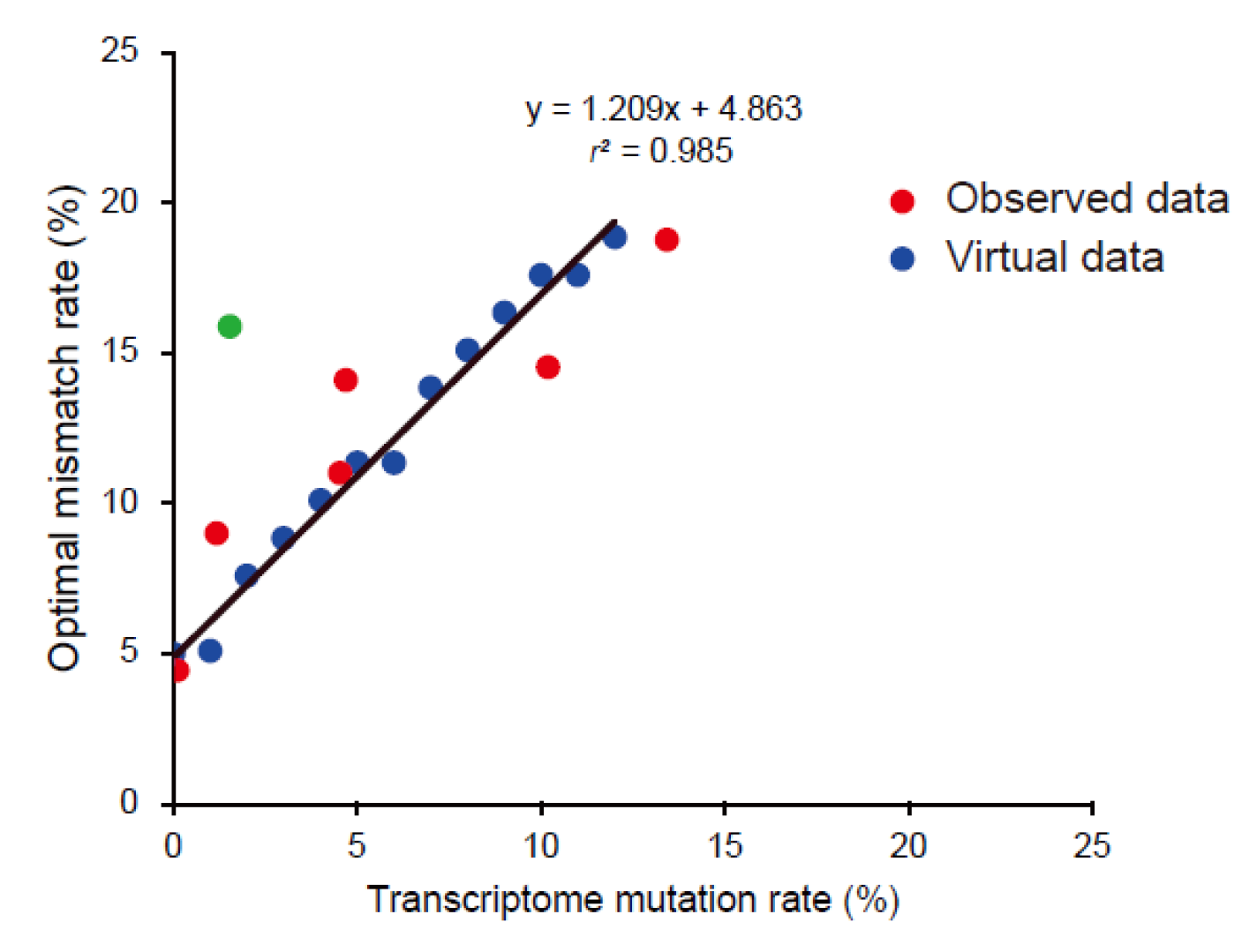
3.5. Building an Optimal Mismatch Rate Model Using Virtual Chicken Genomes
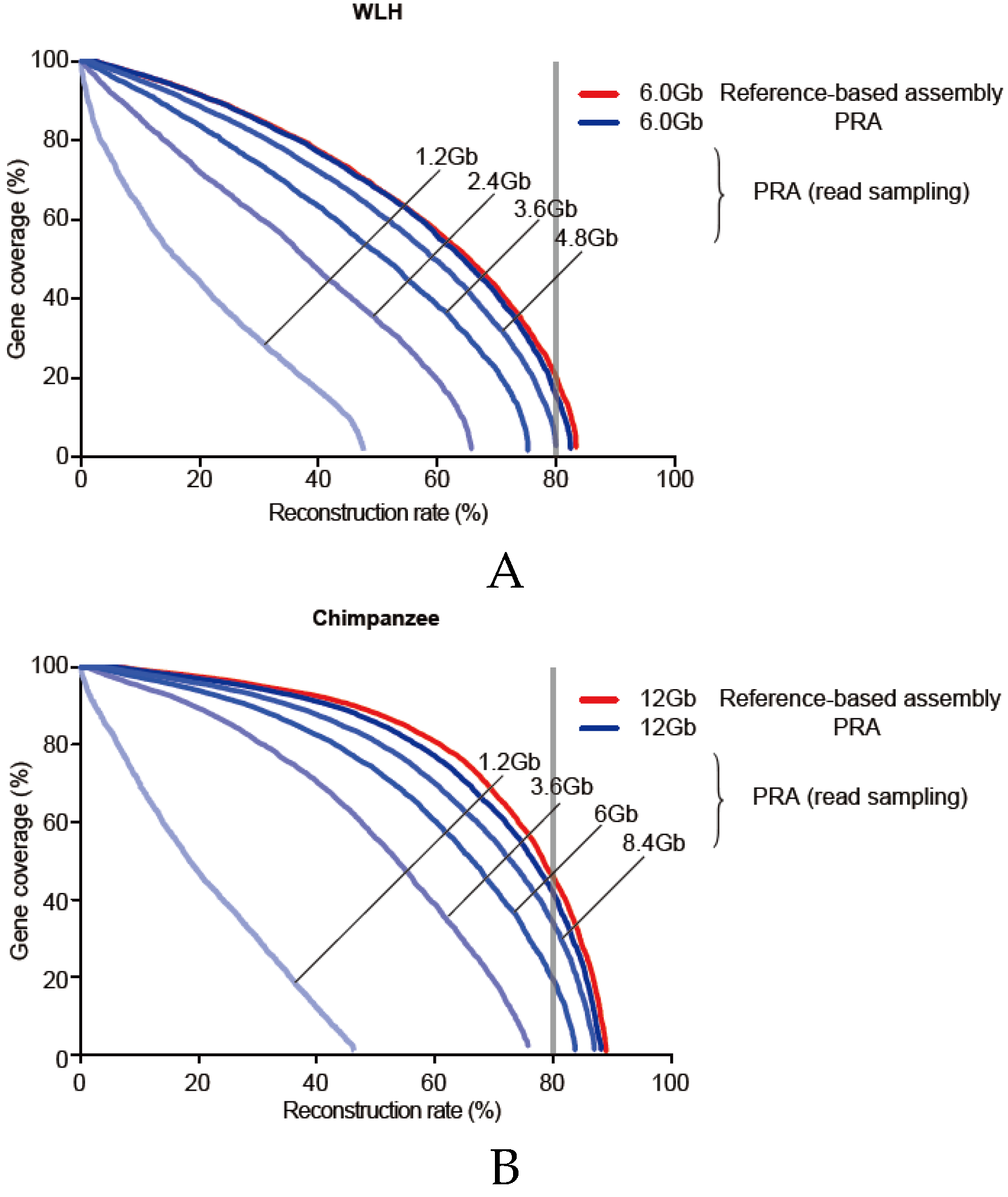
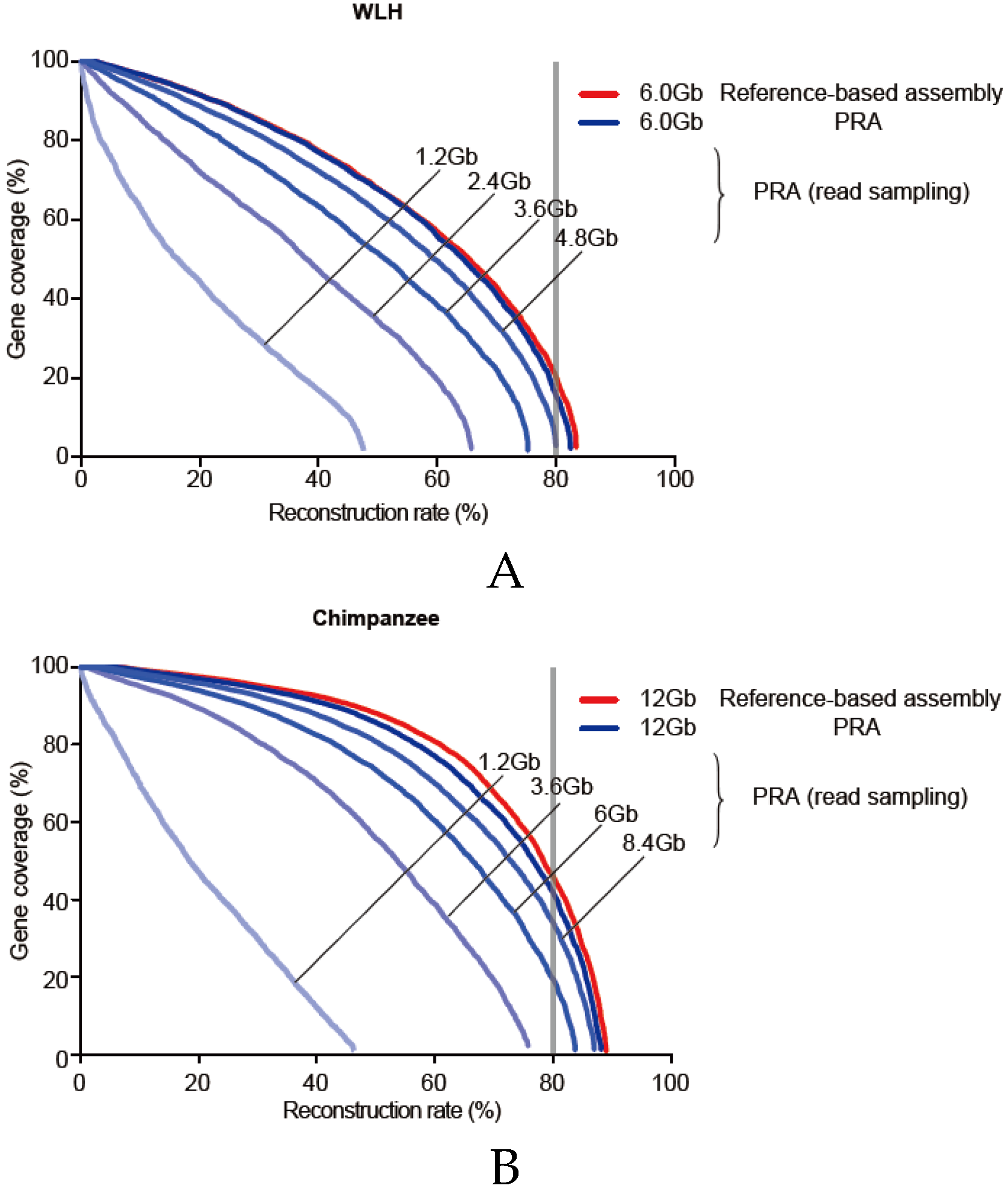
3.6. Required Sequencing Depth for PRA
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.A.; Pirinen, M.; Conrad, D.F.; Lek, M.; Tsang, E.K.; Karczewski, K.J.; Maller, J.B.; Kukurba, K.R.; DeLuca, D.S.; Fromer, M.; et al. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 2015, 348, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Ferreira, P.G.; Reverter, F.; DeLuca, D.S.; Monlong, J.; Sammeth, M.; Young, T.R.; Goldmann, J.M.; Pervouchine, D.D.; Sullivan, T.J.; et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015, 348, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Kircher, M.; He, Z.; Guo, S.; Fairbrother, G.L.; Moreno, C.S.; Khaitovich, P.; Stoneking, M. Evaluating intra- and inter-individual variation in the human placental transcriptome. Genome Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.T.; Marguerat, S.; Watt, S.; Schubert, F.; Wood, V.; Goodhead, I.; Penkett, C.J.; Rogers, J.; Bahler, J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 2008, 453, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Ashtiani, K.; Yang, X.; Derti, A.; Tian, W.; Hao, T.; Lin, C.; Makowski, K.; Shen, L.; Murray, R.R.; Szeto, D.; et al. Isoform discovery by targeted cloning, “deep-well” pooling and parallel sequencing. Nat. Methods 2008, 5, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Hangauer, M.J.; Vaughn, I.W.; McManus, M.T. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013, 9, e1003569. [Google Scholar] [CrossRef] [PubMed]
- Jacquier, A. The complex eukaryotic transcriptome: Unexpected pervasive transcription and novel small RNAs. Nat. Rev. Genet. 2009, 10, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Thomson, N.R. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.B.; Boley, N.; Eisman, R.; May, G.E.; Stoiber, M.H.; Duff, M.O.; Booth, B.W.; Wen, J.; Park, S.; Suzuki, A.M.; et al. Diversity and dynamics of the drosophila transcriptome. Nature 2014, 512, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.B.; Lu, Z.J.; Van Nostrand, E.L.; Cheng, C.; Arshinoff, B.I.; Liu, T.; Yip, K.Y.; Robilotto, R.; Rechtsteiner, A.; Ikegami, K.; et al. Integrative analysis of the caenorhabditis elegans genome by the modencode project. Science 2010, 330, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Fort, A.; Hashimoto, K.; Yamada, D.; Salimullah, M.; Keya, C.A.; Saxena, A.; Bonetti, A.; Voineagu, I.; Bertin, N.; Kratz, A.; et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat. Genet. 2014, 46, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. Gencode: The reference human genome annotation for the encode project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Maudhoo, M.D.; Ren, D.; Gradnigo, J.S.; Gibbs, R.M.; Lubker, A.C.; Moriyama, E.N.; French, J.A.; Norgren, R.B., Jr. De novo assembly of the common marmoset transcriptome from nextgen mRNA sequences. Gigascience 2014. [Google Scholar] [CrossRef] [PubMed]
- Moreton, J.; Dunham, S.P.; Emes, R.D. A consensus approach to vertebrate de novo transcriptome assembly from RNA-seq data: Assembly of the duck (anas platyrhynchos) transcriptome. Front. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Johnson, N.V.; Gross, S.M.; Schnable, J.; Meng, X.; Wang, M.; Coleman-Derr, D.; Lindquist, E.; Wei, C.L.; Kaeppler, S.; et al. A near complete snapshot of the zea mays seedling transcriptome revealed from ultra-deep sequencing. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Bruno, V.M.; Fang, Z.; Meng, X.; Blow, M.; Zhang, T.; Sherlock, G.; Snyder, M.; Wang, Z. Rnnotator: An automated de novo transcriptome assembly pipeline from stranded RNA-seq reads. BMC Genomics 2010. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Li, G.; Liu, J.; Zhang, Y.; Ashby, C.; Liu, D.; Cramer, C.L.; Huang, X. Bridger: A new framework for de novo transcriptome assembly using RNA-seq data. Genome Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Tjaden, B. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. Soapdenovo-trans: De novo transcriptome assembly with short RNA-seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Safikhani, Z.; Sadeghi, M.; Pezeshk, H.; Eslahchi, C. SSP: An interval integer linear programming for de novo transcriptome assembly and isoform discovery of RNA-seq reads. Genomics 2013, 102, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Yassour, M.; Kaplan, T.; Fraser, H.B.; Levin, J.Z.; Pfiffner, J.; Adiconis, X.; Schroth, G.; Luo, S.; Khrebtukova, I.; Gnirke, A.; et al. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 3264–3269. [Google Scholar] [CrossRef] [PubMed]
- Maretty, L.; Sibbesen, J.A.; Krogh, A. Bayesian transcriptome assembly. Genome Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Mangul, S.; Caciula, A.; Al Seesi, S.; Brinza, D.; Mndoiu, I.; Zelikovsky, A. Transcriptome assembly and quantification from ion torrent RNA-seq data. BMC Genomics 2014. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Boley, N.; Stoiber, M.H.; Booth, B.W.; Wan, K.H.; Hoskins, R.A.; Bickel, P.J.; Celniker, S.E.; Brown, J.B. Genome-guided transcript assembly by integrative analysis of RNA sequence data. Nat. Biotechnol. 2014, 32, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, A.M.; Nichols, M.; Burke, T.W.; Ginsburg, G.S.; Lucas, J.E. Comparing reference-based RNA-seq mapping methods for non-human primate data. BMC Genomics 2014. [Google Scholar] [CrossRef] [PubMed]
- Batzoglou, S.; Pachter, L.; Mesirov, J.P.; Berger, B.; Lander, E.S. Human and mouse gene structure: Comparative analysis and application to exon prediction. Genome Res. 2000, 10, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Ureta-Vidal, A.; Ettwiller, L.; Birney, E. Comparative genomics: Genome-wide analysis in metazoan eukaryotes. Nat. Rev. Genet. 2003, 4, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Hardison, R.C. Comparative genomics. PLoS Biol. 2003, 1, e58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Wang, Z.; Collins, J.E.; Andrews, R.M.; Stemple, D.; Gong, Z. Comparative transcriptome analyses indicate molecular homology of zebrafish swimbladder and mammalian lung. PLoS ONE 2011, 6, e24019. [Google Scholar] [CrossRef] [PubMed]
- Wellcome Trust Sanger Institute. Ensembl. Available online: http://www.ensembl.org/ (accessed on 24 December 2015).
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FASTQ Files. Available online: https://github.com/najoshi/sickle/ (accessed on 24 December 2015).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMB Net. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information, U.S. National Library of Medicine. NCBI Nucleotide. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/nucleotide/ (accessed on 24 December 2015).
- Roth, A.C.; Gonnet, G.H.; Dessimoz, C. Algorithm of oma for large-scale orthology inference. BMC Bioinform. 2008. [Google Scholar] [CrossRef] [PubMed]
- Dessimoz Lab at UCL, CBRG at ETH Zurich. Orthologous Matrix. Available online: http://omabrowser.org/oma/home/ (accessed on 24 December 2015).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal w: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. Tophat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information, U.S. National Library of Medicine. NCBI Genome. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/assembly/organism/ (accessed on 24 December 2015).




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, K.; Jeong, H.; Nam, J.-W. Pseudo-Reference-Based Assembly of Vertebrate Transcriptomes. Genes 2016, 7, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7030010
Nam K, Jeong H, Nam J-W. Pseudo-Reference-Based Assembly of Vertebrate Transcriptomes. Genes. 2016; 7(3):10. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7030010
Chicago/Turabian StyleNam, Kyoungwoo, Heesu Jeong, and Jin-Wu Nam. 2016. "Pseudo-Reference-Based Assembly of Vertebrate Transcriptomes" Genes 7, no. 3: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7030010