
Remarkable Single Atom Catalyst of Transition Metal (Fe, Co & Ni) Doped on C2N Surface for Hydrogen Dissociation Reaction
 , , and
, , and
Abstract
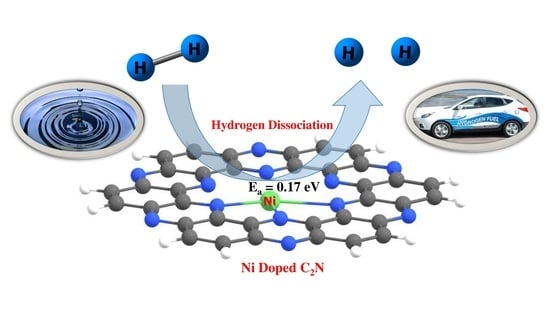
:
1. Introduction
2. Computational Methodology
3. Results and Discussion
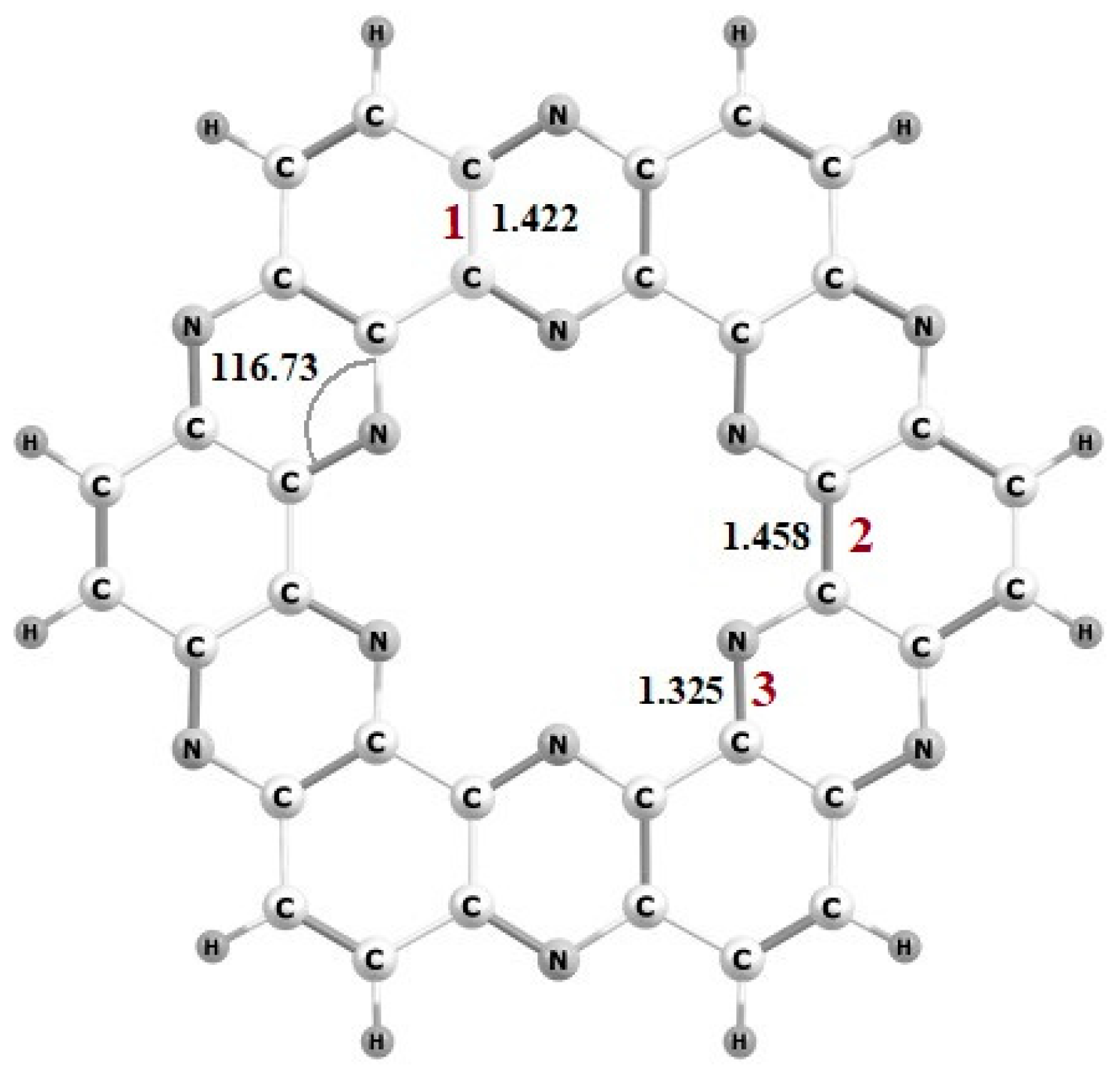
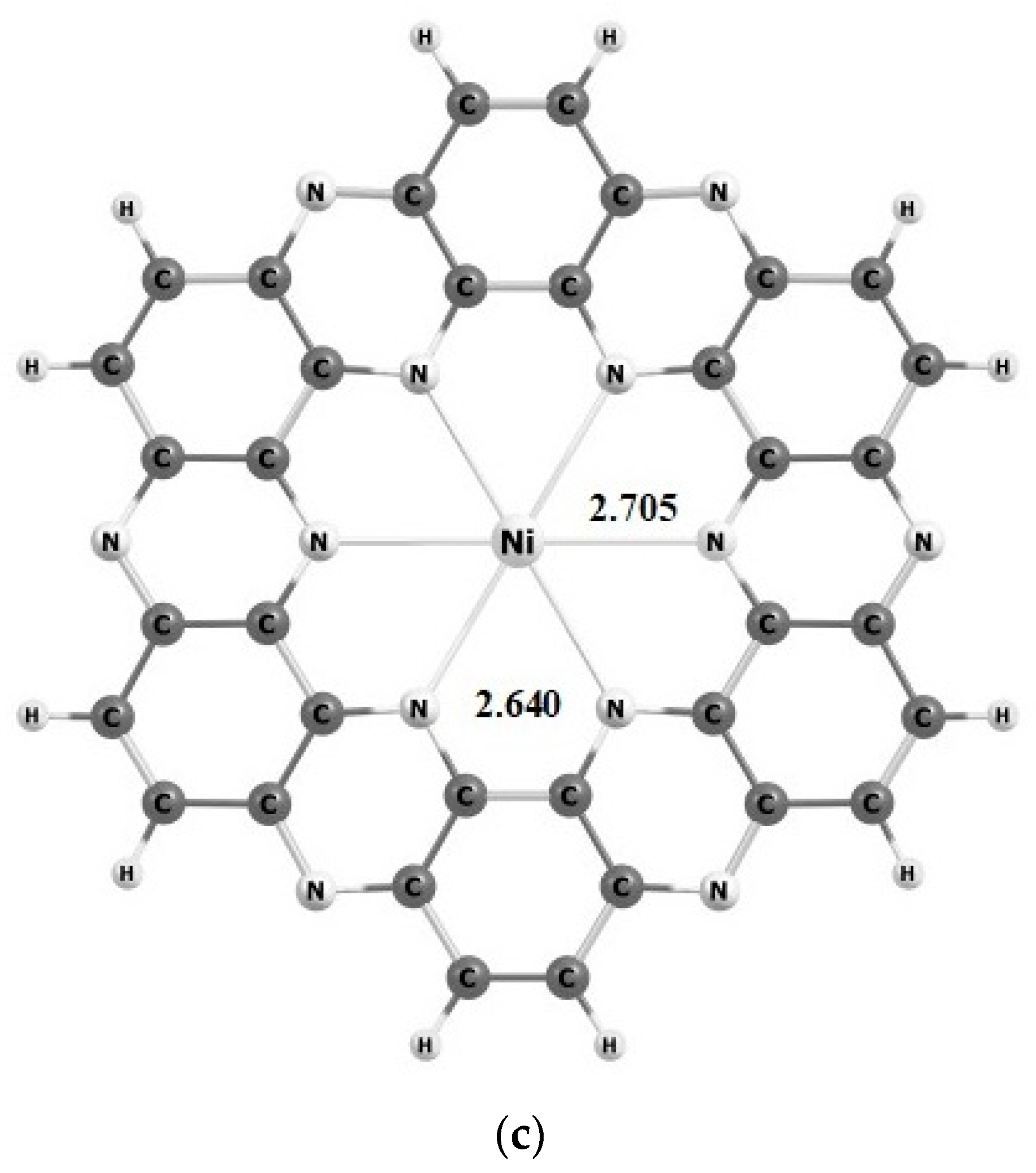
Geometries and Electronic Properties
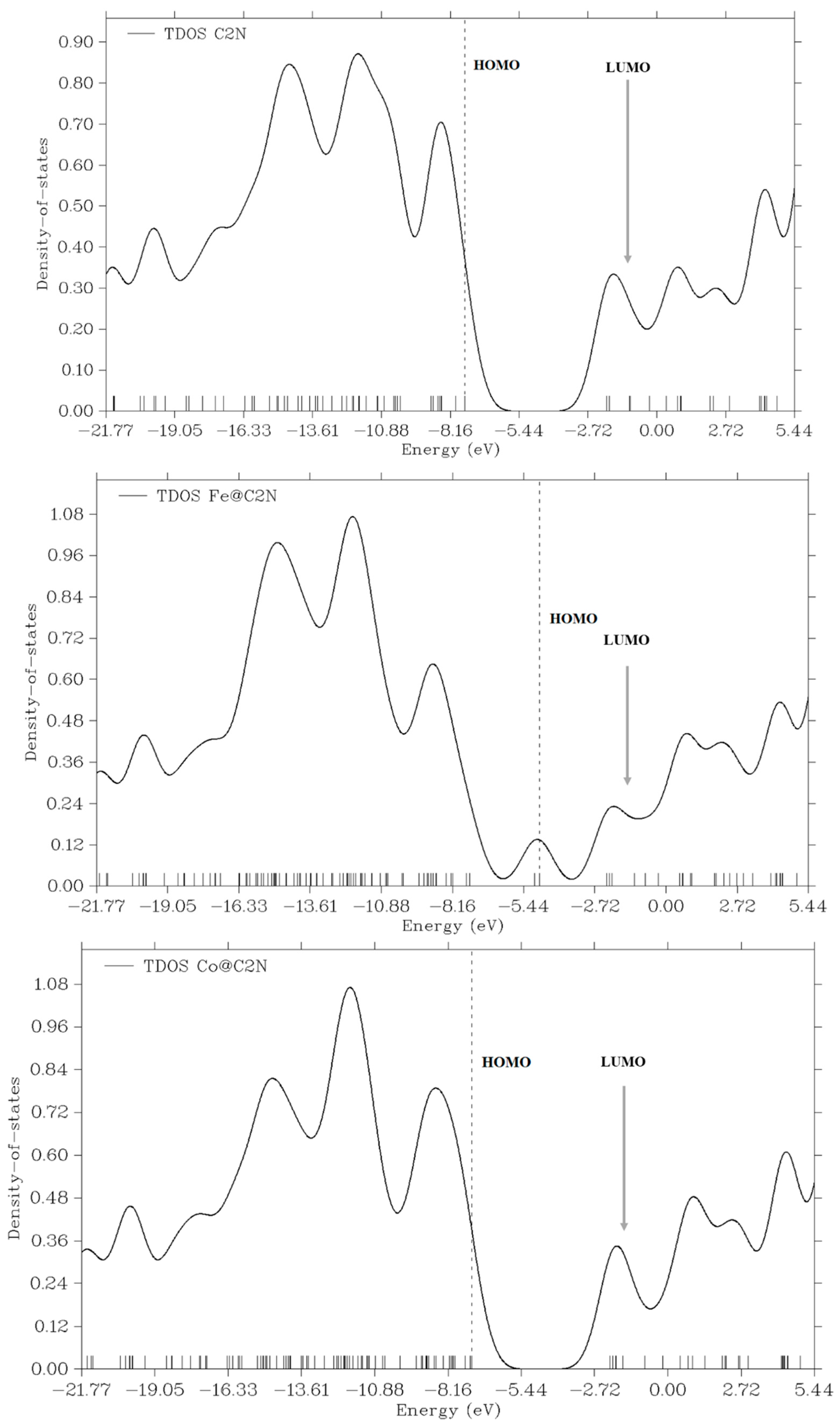
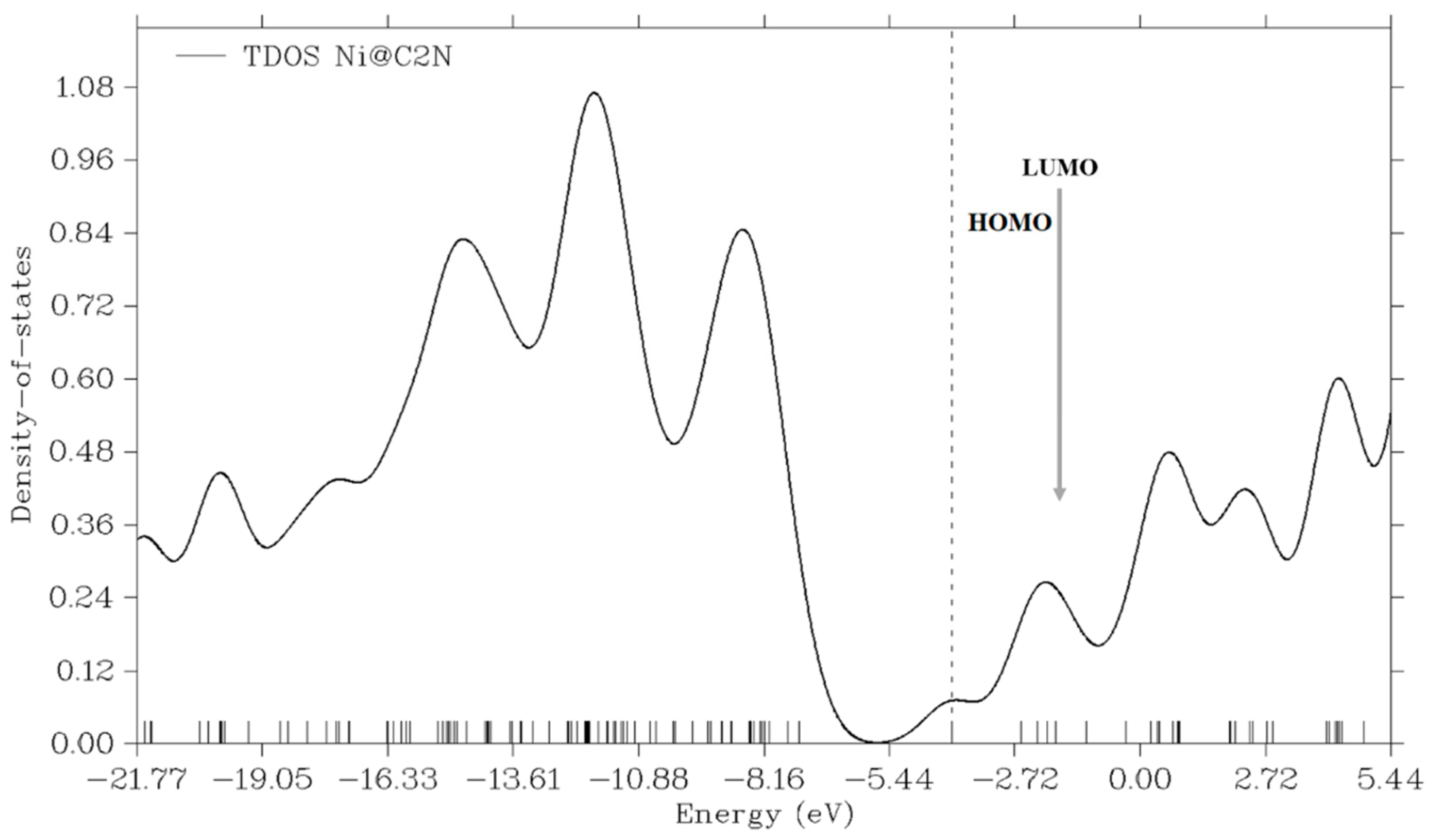
4. HOMO-LUMO and DOS Analysis
5. Natural Bond Orbital (NBO) Analysis
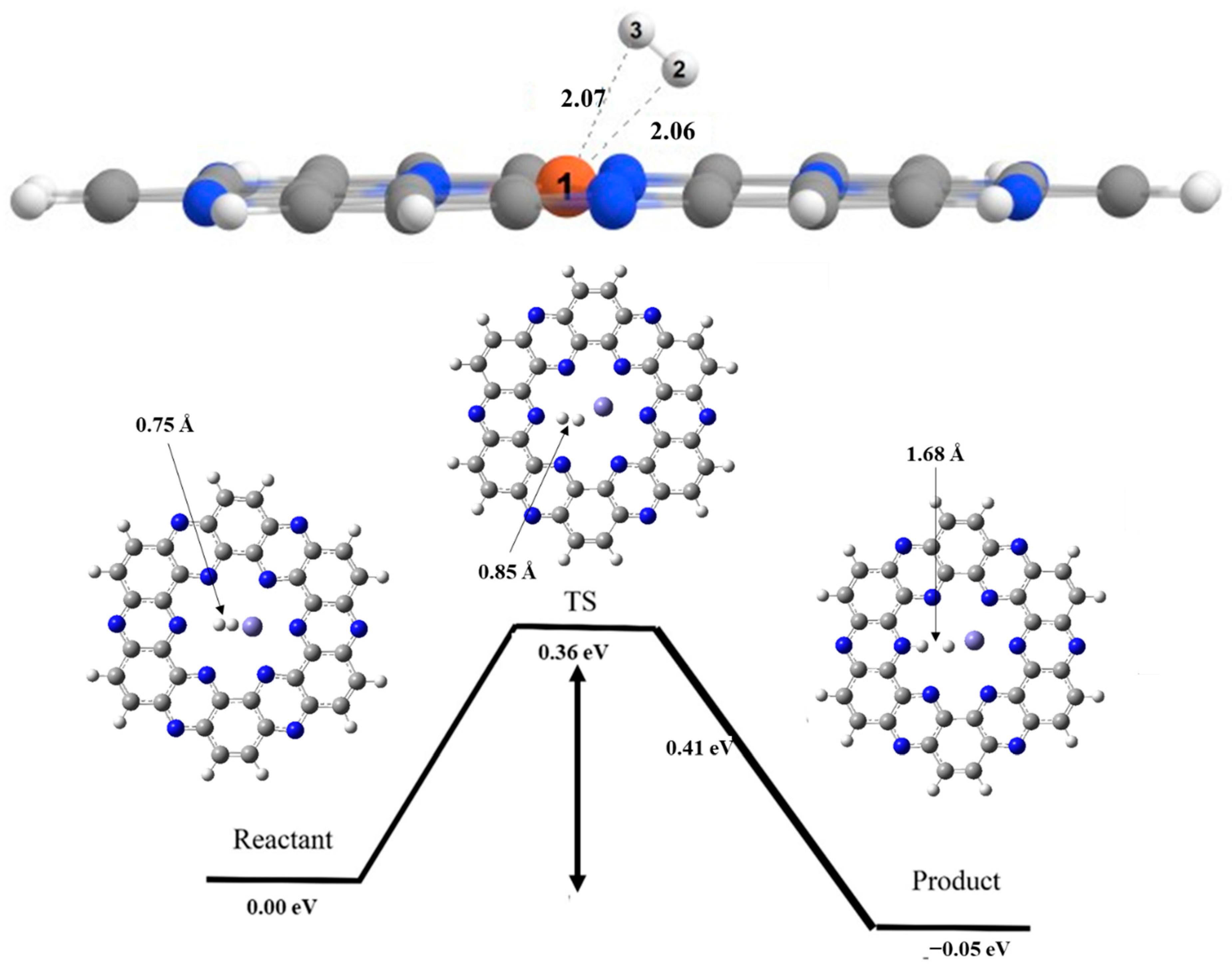
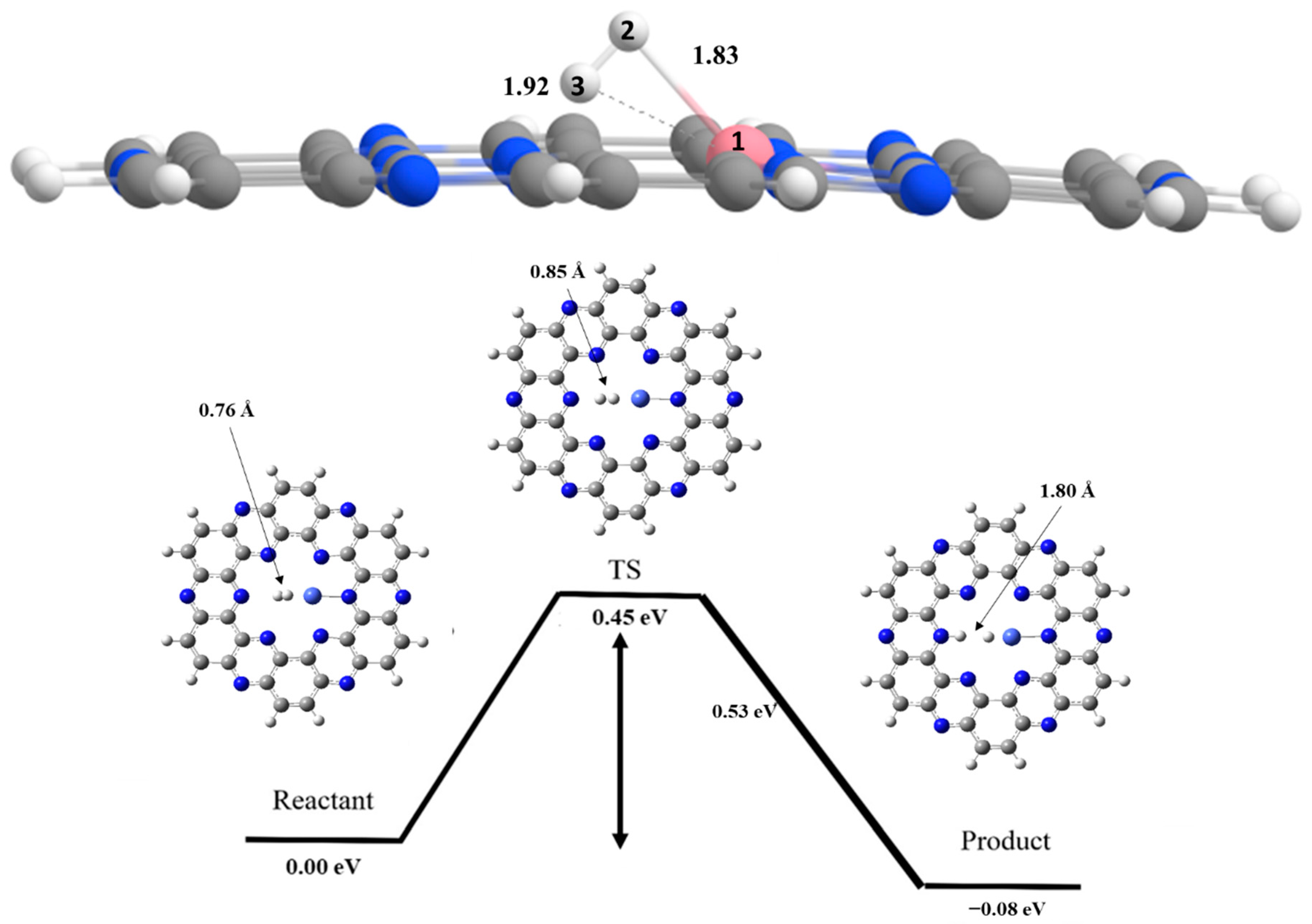
6. Hydrogen Dissociation Reaction on Iron Doped C2N Surface
7. Hydrogen Dissociation Reaction on Cobalt Doped C2N Surface
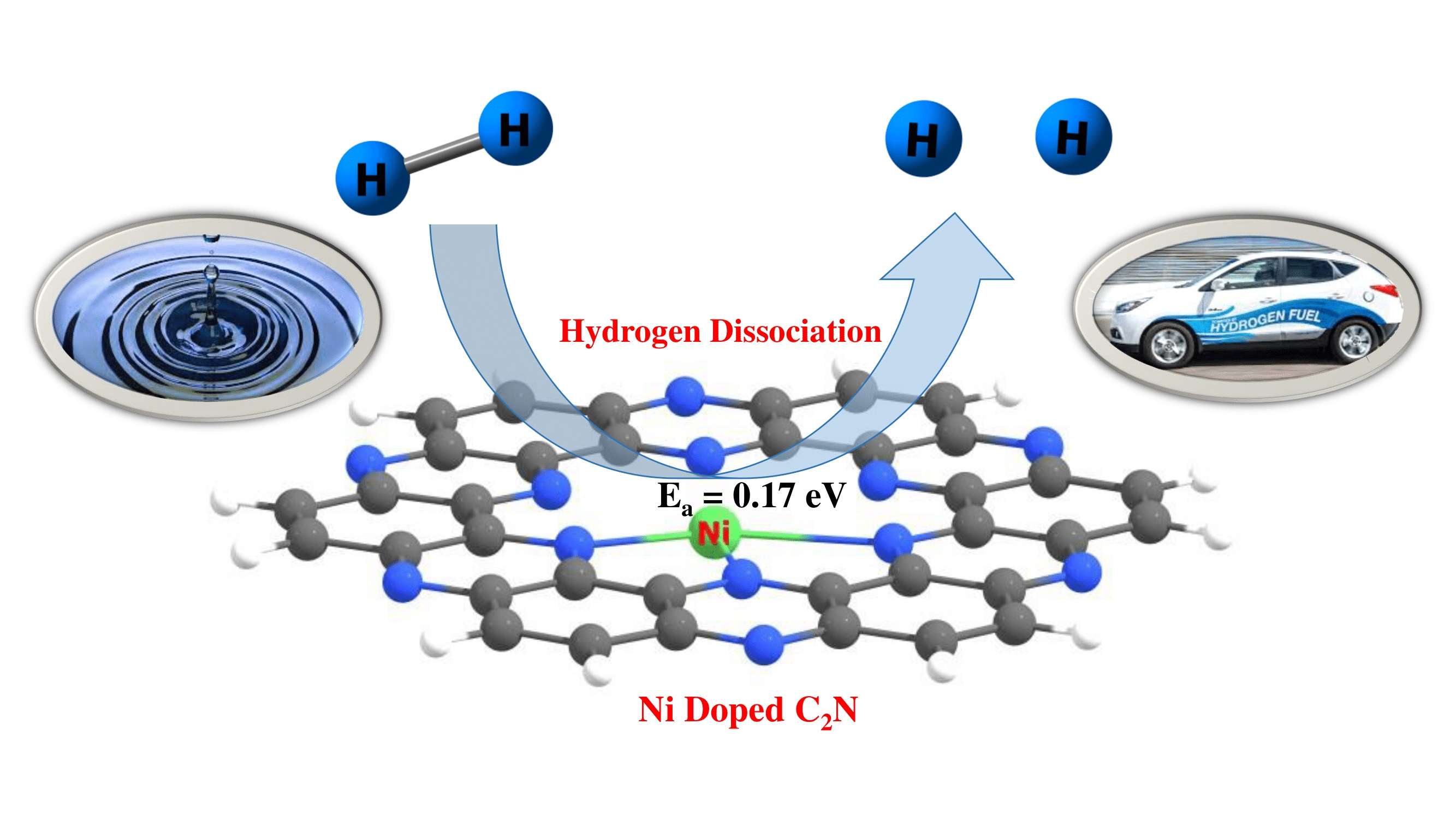
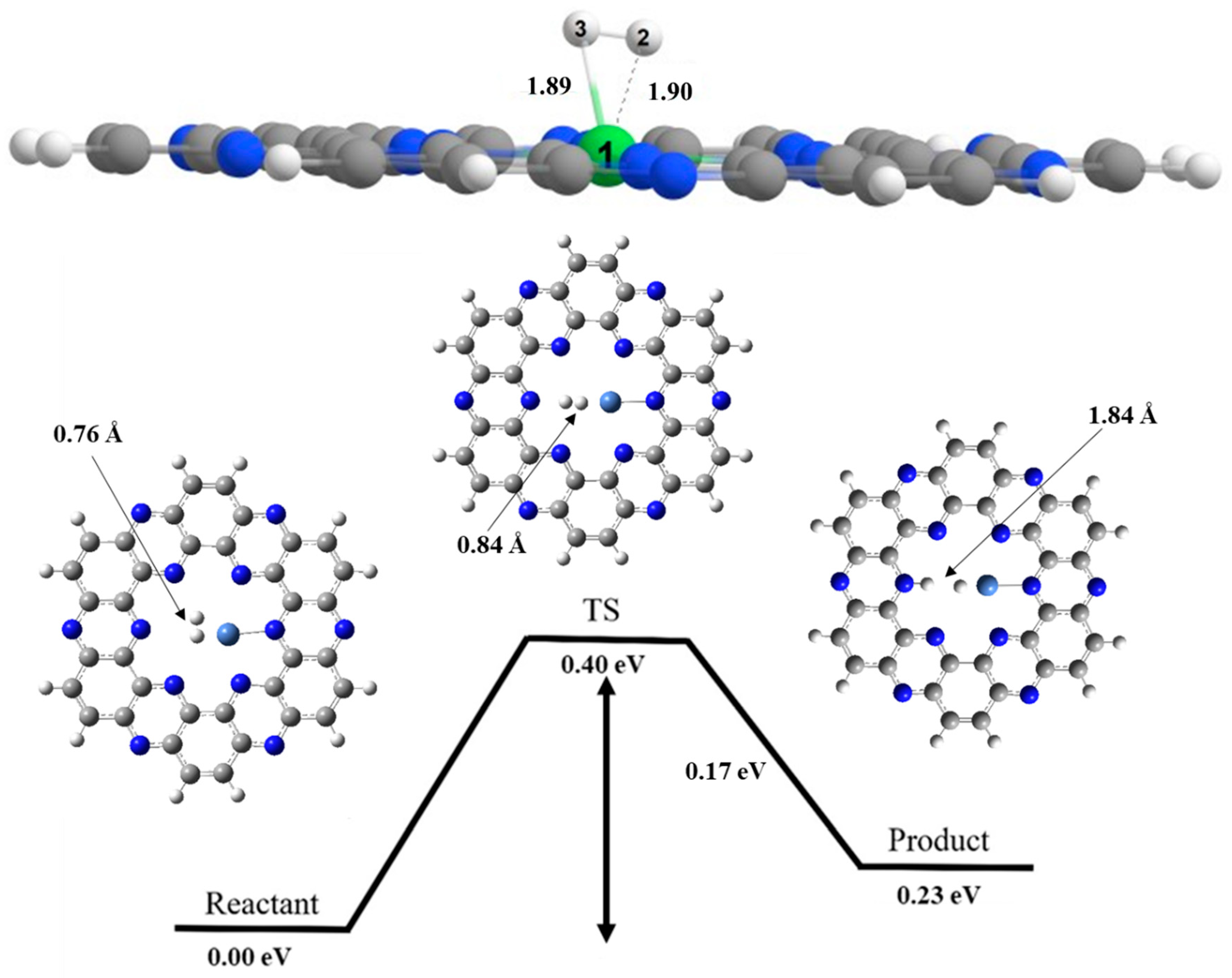
8. Hydrogen Dissociation Reaction on Nickel Doped C2N Surface
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Hagen, J. Industrial Catalysis: A Practical Approach; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Li, X.; Yang, X.; Huang, Y.; Zhang, T.; Liu, B. Supported Noble-Metal Single Atoms for Heterogeneous Catalysis. Adv. Mater. 2019, 31, 1902031. [Google Scholar] [CrossRef] [PubMed]
- Bigall, N.C.; Reitzig, M.; Naumann, W.; Simon, P.; van Pée, K.H.; Eychmüller, A. Fungal templates for noble-metal nanoparticles and their application in catalysis. Angew. Chem. 2008, 120, 7994–7997. [Google Scholar] [CrossRef]
- De Beer, M.; Kunene, A.; Nabaho, D.; Claeys, M.; Van Steen, E. Technical and economic aspects of promotion of cobalt-based Fischer-Tropsch catalysts by noble metals—A review. J. South. Afr. Inst. Min. Metall. 2014, 114, 157–165. [Google Scholar]
- Liu, D.; Barbar, A.; Najam, T.; Javed, M.S.; Shen, J.; Tsiakaras, P.; Cai, X. Single noble metal atoms doped 2D materials for catalysis applications. Appl. Catal. B Environ. 2021, 297, 120389. [Google Scholar] [CrossRef]
- Liang, S.; Hao, C.; Shi, Y. The power of single-atom catalysis. ChemCatChem 2015, 7, 2559–2567. [Google Scholar] [CrossRef]
- Chen, F.; Jiang, X.; Zhang, L.; Lang, R.; Qiao, B. Single-atom catalysis: Bridging the homo-and heterogeneous catalysis. Chin. J. Catal. 2018, 39, 893–898. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, S.; Chen, C.; Peng, Q.; Wang, D.; Li, Y. Single-atom catalysts: Synthetic strategies and electrochemical applications. Joule 2018, 2, 1242–1264. [Google Scholar] [CrossRef] [Green Version]
- Cheng, N.; Sun, X. Single atom catalyst by atomic layer deposition technique. Chin. J. Catal. 2017, 38, 1508–1514. [Google Scholar] [CrossRef]
- Righi, G.; Magri, R.; Selloni, A. H2 dissociation on noble metal single atom catalysts adsorbed on and doped into CeO2 (111). J. Phys. Chem. C 2019, 123, 9875–9883. [Google Scholar] [CrossRef]
- Thiel, W. Computational catalysis—Past, present, and future. Angew. Chem. Int. Ed. 2014, 53, 8605–8613. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Huang, J.; Wang, Z.-J.; Liu, X.-X.; Li, J.; Li, Z.-R. Superalkali-alkalide ion pairs δ+(M-HMHC)-M’ δ−(M, M’ = Li, Na and K) serving as high-performance NLO molecular materials. J. Mol. Liq. 2021, 349, 118101. [Google Scholar] [CrossRef]
- Eftekhari, A.; Fang, B. Electrochemical hydrogen storage: Opportunities for fuel storage, batteries, fuel cells, and supercapacitors. Int. J. Hydrog. Energy 2017, 42, 25143–25165. [Google Scholar] [CrossRef]
- Zang, G.; Sun, P.; Elgowainy, A.A.; Bafana, A.; Wang, M. Performance and cost analysis of liquid fuel production from H2 and CO2 based on the Fischer-Tropsch process. J. CO2 Util. 2021, 46, 101459. [Google Scholar] [CrossRef]
- Kyriakou, V.; Garagounis, I.; Vourros, A.; Vasileiou, E.; Stoukides, M. An electrochemical haber-bosch process. Joule 2020, 4, 142–158. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Xiao, M.; Gao, L.; Wang, Y.; Wang, X.; Zhu, J.; Jin, Z.; Liu, C.; Chen, H.; Li, G.; Ge, J.; et al. Engineering energy level of metal center: Ru single-atom site for efficient and durable oxygen reduction catalysis. J. Am. Chem. Soc. 2019, 141, 19800–19806. [Google Scholar] [CrossRef]
- Guo, Y.; Lang, R.; Qiao, B. Highlights of major progress on single-atom catalysis in 2017. Catalysts 2019, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, T.K.; Nair, N.N. Rh1/γ-Al2O3 Single-Atom Catalysis of O2 Activation and CO Oxidation: Mechanism, Effects of Hydration, Oxidation State, and Cluster Size. ChemCatChem 2013, 5, 1811–1821. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Y.; Chu, M.; Duan, T.; Meng, C.; Han, Y. Defect stabilized gold atoms on graphene as potential catalysts for ethylene epoxidation: A first-principles investigation. Catal. Sci. Technol. 2016, 6, 1632–1641. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, G.S. Single-atom catalysis: How structure influences catalytic performance. Catal. Lett. 2019, 149, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Zhang, L.; Doyle-Davis, K.; Sun, X. Single-atom catalysts: From design to application. Electrochem. Energy Rev. 2019, 2, 539–573. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.-X.; Yang, X.-F.; Wang, A.; Zhang, T.; Li, J. Theoretical investigations of non-noble metal single-atom catalysis: Ni1/FeOx for CO oxidation. Catal. Sci. Technol. 2016, 6, 6886–6892. [Google Scholar] [CrossRef]
- Sun, T.; Xu, L.; Wang, D.; Li, Y. Metal organic frameworks derived single atom catalysts for electrocatalytic energy conversion. Nano Res. 2019, 12, 2067–2080. [Google Scholar] [CrossRef]
- Ma, D.; Li, T.; Wang, Q.; Yang, G.; He, C.; Ma, B.; Lu, Z. Graphyne as a promising substrate for the noble-metal single-atom catalysts. Carbon 2015, 95, 756–765. [Google Scholar] [CrossRef]
- Ren, S.; Yu, Q.; Yu, X.; Rong, P.; Jiang, L.; Jiang, J. Graphene-supported metal single-atom catalysts: A concise review. Sci. China Mater. 2020, 63, 903–920. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Wang, S.; Wang, Z.; Liu, K.; Li, H.; Liu, H.; Hu, J.; Xu, X.; Li, H.; Liu, M. Graphitic carbon nitride based single-atom photocatalysts. Front. Phys. 2020, 15, 33201. [Google Scholar] [CrossRef]
- Papa, V.; Cao, Y.; Spannenberg, A.; Junge, K.; Beller, M. Development of a practical non-noble metal catalyst for hydrogenation of N-heteroarenes. Nat. Catal. 2020, 3, 135–142. [Google Scholar] [CrossRef]
- Franco, F.; Rettenmaier, C.; Jeon, H.S.; Cuenya, B.R. Transition metal-based catalysts for the electrochemical CO2 reduction: From atoms and molecules to nanostructured materials. Chem. Soc. Rev. 2020, 49, 6884–6946. [Google Scholar] [CrossRef]
- Yan, H.; Lv, H.; Yi, H.; Liu, W.; Xia, Y.; Huang, X.; Huang, W.; Wei, S.; Wu, X.; Lu, J. Understanding the underlying mechanism of improved selectivity in pd1 single-atom catalyzed hydrogenation reaction. J. Catal. 2018, 366, 70–79. [Google Scholar] [CrossRef]
- Mahmood, J.; Lee, E.K.; Jung, M.; Shin, D.; Jeon, I.-Y.; Jung, S.-M.; Choi, H.-J.; Seo, J.-M.; Bae, S.-Y.; Sohn, S.-D.; et al. Nitrogenated holey two-dimensional structures. Nat. Commun. 2015, 6, 6486. [Google Scholar] [CrossRef] [PubMed]
- Yar, M.; Shah, A.B.; Hashmi, M.A.; Ayub, K. Selective detection and removal of picric acid by C2N surface from a mixture of nitro-explosives. New J. Chem. 2020, 44, 18646–18655. [Google Scholar] [CrossRef]
- Panigrahi, P.; Desai, M.; Talari, M.K.; Bae, H.; Lee, H.; Ahuja, R.; Hussain, T. Selective decoration of nitrogenated holey graphene (C2N) with titanium clusters for enhanced hydrogen storage application. Int. J. Hydrog. Energy 2021, 46, 7371–7380. [Google Scholar] [CrossRef]
- Li, X.; Zhong, W.; Cui, P.; Li, J.; Jiang, J. Design of efficient catalysts with double transition metal atoms on C2N layer. J. Phys. Chem. Lett. 2016, 7, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Zhao, L.; Liu, S.; Duan, G.; Perez-Aguilar, J.M.; Luo, J.; Li, W.; Zhou, R. Orientational binding of DNA guided by the C2N template. ACS Nano 2017, 11, 3198–3206. [Google Scholar] [CrossRef]
- Liang, Z.; Yang, D.; Tang, P.; Zhang, C.; Jacas Biendicho, J.; Zhang, Y.; Llorca, J.; Wang, X.; Li, J.; Heggen, M. Atomically dispersed Fe in a C2N based catalyst as a sulfur host for efficient lithium–sulfur batteries. Adv. Energy Mater. 2021, 11, 2003507. [Google Scholar] [CrossRef]
- Barrio, J.; Pedersen, A.; Feng, J.; Sarma, S.C.; Wang, M.; Li, A.Y.; Yadegari, H.; Luo, H.; Ryan, M.P.; Titirici, M.-M.; et al. Metal coordination in C2N-like materials towards dual atom catalysts for oxygen reduction. J. Mater. Chem. A 2022, 10, 6023–6030. [Google Scholar] [CrossRef]
- He, B.; Shen, J.; Tian, Z. Iron-embedded C2N monolayer: A promising low-cost and high-activity single-atom catalyst for CO oxidation. Phys. Chem. Chem. Phys. 2016, 18, 24261–24269. [Google Scholar] [CrossRef]
- Mahmood, J.; Li, F.; Kim, C.; Choi, H.-J.; Gwon, O.; Jung, S.-M.; Seo, J.-M.; Cho, S.-J.; Ju, Y.-W.; Jeong, H.Y.; et al. Fe@C2N: A highly-efficient indirect-contact oxygen reduction catalyst. Nano Energy 2018, 44, 304–310. [Google Scholar] [CrossRef]
- Ma, J.; Gong, H.; Zhang, T.; Yu, H.; Zhang, R.; Liu, Z.; Yang, G.; Sun, H.; Tang, S.; Qiu, Y. Hydrogenation of CO2 to formic acid on the single atom catalysis Cu/C2N: A first principles study. Appl. Surf. Sci. 2019, 488, 1–9. [Google Scholar] [CrossRef]
- Zhong, W.; Zhang, G.; Zhang, Y.; Jia, C.; Yang, T.; Ji, S.; Prezhdo, O.V.; Yuan, J.; Luo, Y.; Jiang, J. Enhanced activity of C2N-supported single Co atom catalyst by single atom promoter. J. Phys. Chem. Lett. 2019, 10, 7009–7014. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wang, Q.; Yan, X.; Zhang, X.; He, C.; Zhou, D.; Tang, Y.; Lu, Z.; Yang, Z. 3d transition metal embedded C2N monolayers as promising single-atom catalysts: A first-principles study. Carbon 2016, 105, 463–473. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Das, T.; Banerjee, P.; Thapa, R.; Das, G. Electron doped C2N monolayer as efficient noble metal-free catalysts for CO oxidation. Appl. Surf. Sci. 2017, 418, 92–98. [Google Scholar] [CrossRef]
- Egorova, K.S.; Ananikov, V.P. Which metals are green for catalysis? Comparison of the toxicities of Ni, Cu, Fe, Pd, Pt, Rh, and Au salts. Angew. Chem. Int. Ed. 2016, 55, 12150–12162. [Google Scholar]
- Du, Z.; Chen, X.; Hu, W.; Chuang, C.; Xie, S.; Hu, A.; Yan, W.; Kong, X.; Wu, X.; Ji, H.; et al. Cobalt in nitrogen-doped graphene as single-atom catalyst for high-sulfur content lithium–sulfur batteries. J. Am. Chem. Soc. 2019, 141, 3977–3985. [Google Scholar] [CrossRef]
- Bing, Q.; Liu, W.; Yi, W.; Liu, J.-Y. Ni anchored C2N monolayers as low-cost and efficient catalysts for hydrogen production from formic acid. J. Power Sources 2019, 413, 399–407. [Google Scholar] [CrossRef]
- Qin, G.; Cui, Q.; Yun, B.; Sun, L.; Du, A.; Sun, Q. High capacity and reversible hydrogen storage on two dimensional C2N monolayer membrane. Int. J. Hydrog. Energy 2018, 43, 9895–9901. [Google Scholar] [CrossRef]
- Li, X.; Cui, P.; Zhong, W.; Li, J.; Wang, X.; Wang, Z.; Jiang, J. Graphitic carbon nitride supported single-atom catalysts for efficient oxygen evolution reaction. Chem. Commun. 2016, 52, 13233–13236. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Hohenstein, E.G.; Chill, S.T.; Sherrill, C.D. Assessment of the Performance of the M05−2X and M06−2X Exchange-Correlation Functionals for Noncovalent Interactions in Biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef]
- Tian, Z.; López-Salas, N.; Liu, C.; Liu, T.; Antonietti, M. C2N: A Class of Covalent Frameworks with Unique Properties. Adv. Sci. 2020, 7, 2001767. [Google Scholar] [CrossRef] [PubMed]
- Badran, H.; Eid, K.M.; Ammar, H. DFT and TD-DFT studies of halogens adsorption on cobalt-doped porphyrin: Effect of the external electric field. Results Phys. 2021, 23, 103964. [Google Scholar] [CrossRef]
- Yang, T.; Fukuda, R.; Hosokawa, S.; Tanaka, T.; Sakaki, S.; Ehara, M. A theoretical investigation on CO oxidation by single-atom catalysts M1/γ-Al2O3 (M= Pd, Fe, Co, and Ni). ChemCatChem 2017, 9, 1222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, X.; Liu, C.; Guo, J.; Ning, H. Hydrogen adsorption and dissociation on nickel-adsorbed and-substituted Mg17Al12 (100) surface: A density functional theory study. Int. J. Hydrog. Energy 2018, 43, 793–800. [Google Scholar] [CrossRef]
- Du, A.; Smith, S.C.; Yao, X.; Lu, G. The role of Ti as a catalyst for the dissociation of hydrogen on a Mg (0001) surface. J. Phys. Chem. B 2005, 109, 18037–18041. [Google Scholar] [CrossRef]
- Sun, K.; Kohyama, M.; Tanaka, S.; Takeda, S. A study on the mechanism for H2 dissociation on Au/TiO2 catalysts. J. Phys. Chem. C 2014, 118, 1611–1617. [Google Scholar] [CrossRef]
- Trivedi, R.; Bandyopadhyay, D. Study of adsorption and dissociation pathway of H2 molecule on MgnRh (n = 1–10) clusters: A first principle investigation. Int. J. Hydrog. Energy 2016, 41, 20113–20121. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
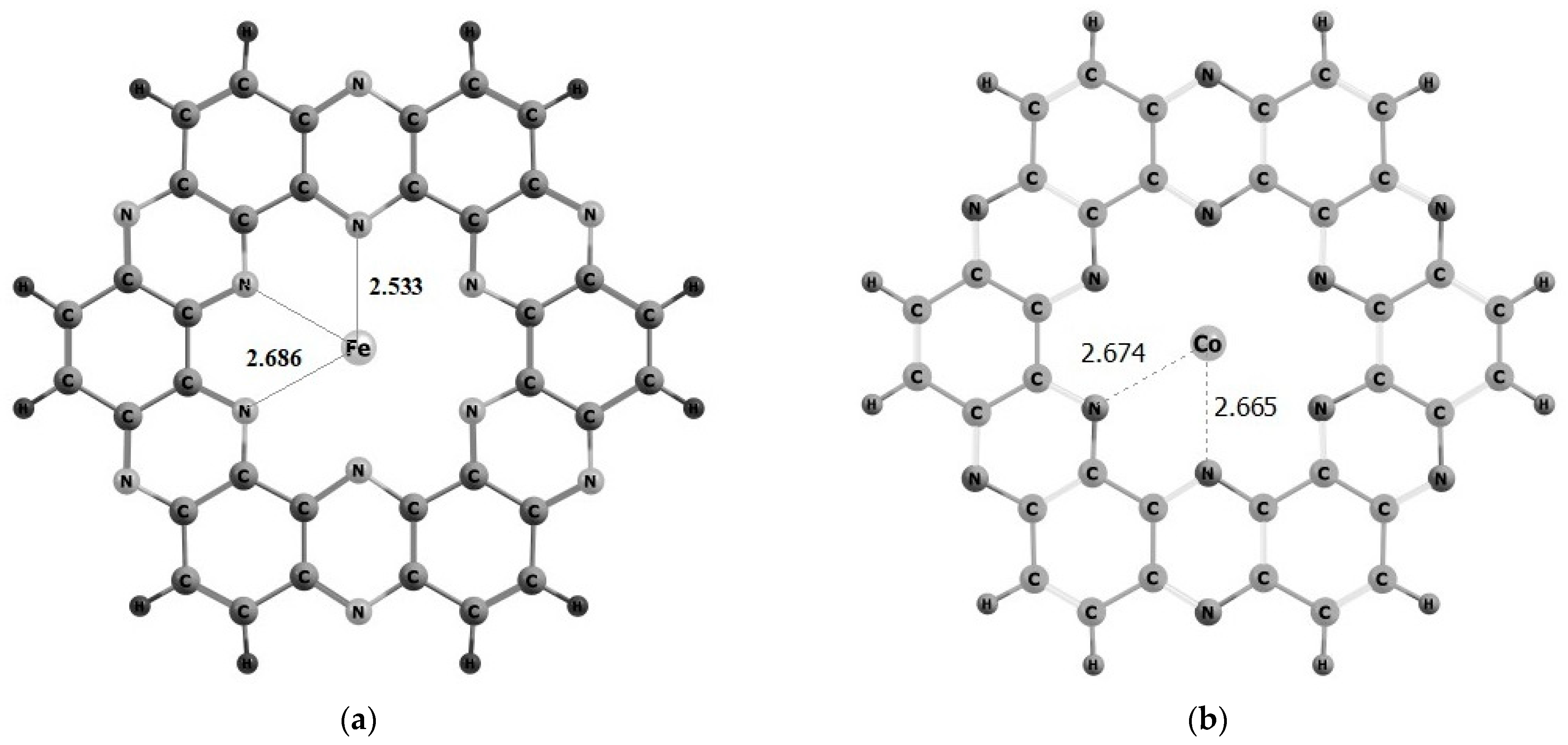
| M@C2N | Eint | M-N | NBO | HOMO | LUMO | EH-L |
|---|---|---|---|---|---|---|
| Fe@C2N | −3.19 | 2.53, 2.69 | 1.43 | −4.84 | −2.29 | 2.56 |
| Co@C2N | −1.42 | 2.66, 2.67 | 0.92 | −7.27 | −2.15 | 5.11 |
| Ni@C2N | −2.51 | 2.64, 2.71 | 0.74 | −4.08 | −2.59 | 1.50 |
| C2N | -- | -- | -- | −7.59 | −1.99 | 5.61 |
| Reaction Energies | Fe@C2N | Co@C2N | Ni@C2N | |
|---|---|---|---|---|
| Ea | 0.36 | 0.45 | 0.40 | |
| ΔE | −0.05 | −0.08 | 0.23 | |
| H—H Bond length | B.LR | 0.76 | 0.75 | 0.76 |
| B.LTS | 0.93 | 0.85 | 0.84 | |
| B.LP | 1.68 | 1.80 | 1.84 | |
| Ni—H Bond length | B.LR | 2.06 | 1.83 | 1.89 |
| B.LTS | 1.78 | 1.76 | 1.75 | |
| B.LP | 1.67 | 1.60 | 1.56 | |
| Surfaces | Dissociation Barrier | References |
|---|---|---|
| Ni adsorbed Mg17Al12 surface | Mg16NiAl12 0.82 eV Mg15Ni2Al12 0.53 eV | [55] |
| Ti doped Mg Surface | 0.35 eV | [56] |
| Au/TiO2 | 0.54 eV | [57] |
| Mg9Rh cluster | 0.63 eV | [58] |
| Fe@C2N | 0.36 eV | This work |
| Co@C2N | 0.45 eV | -- |
| Ni@C2N | 0.40 eV | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, A.B.; Sarfaraz, S.; Yar, M.; Sheikh, N.S.; Hammud, H.H.; Ayub, K. Remarkable Single Atom Catalyst of Transition Metal (Fe, Co & Ni) Doped on C2N Surface for Hydrogen Dissociation Reaction. Nanomaterials 2023, 13, 29. https://0-doi-org.brum.beds.ac.uk/10.3390/nano13010029
Shah AB, Sarfaraz S, Yar M, Sheikh NS, Hammud HH, Ayub K. Remarkable Single Atom Catalyst of Transition Metal (Fe, Co & Ni) Doped on C2N Surface for Hydrogen Dissociation Reaction. Nanomaterials. 2023; 13(1):29. https://0-doi-org.brum.beds.ac.uk/10.3390/nano13010029
Chicago/Turabian StyleShah, Ahmed Bilal, Sehrish Sarfaraz, Muhammad Yar, Nadeem S. Sheikh, Hassan H. Hammud, and Khurshid Ayub. 2023. "Remarkable Single Atom Catalyst of Transition Metal (Fe, Co & Ni) Doped on C2N Surface for Hydrogen Dissociation Reaction" Nanomaterials 13, no. 1: 29. https://0-doi-org.brum.beds.ac.uk/10.3390/nano13010029