
Elucidating miRNA Function in Cancer Biology via the Molecular Genetics’ Toolbox

 and
and 
Abstract
:1. MicroRNA: A Brief Introduction
2. Mechanisms of microRNA Dysregulation in Cancer
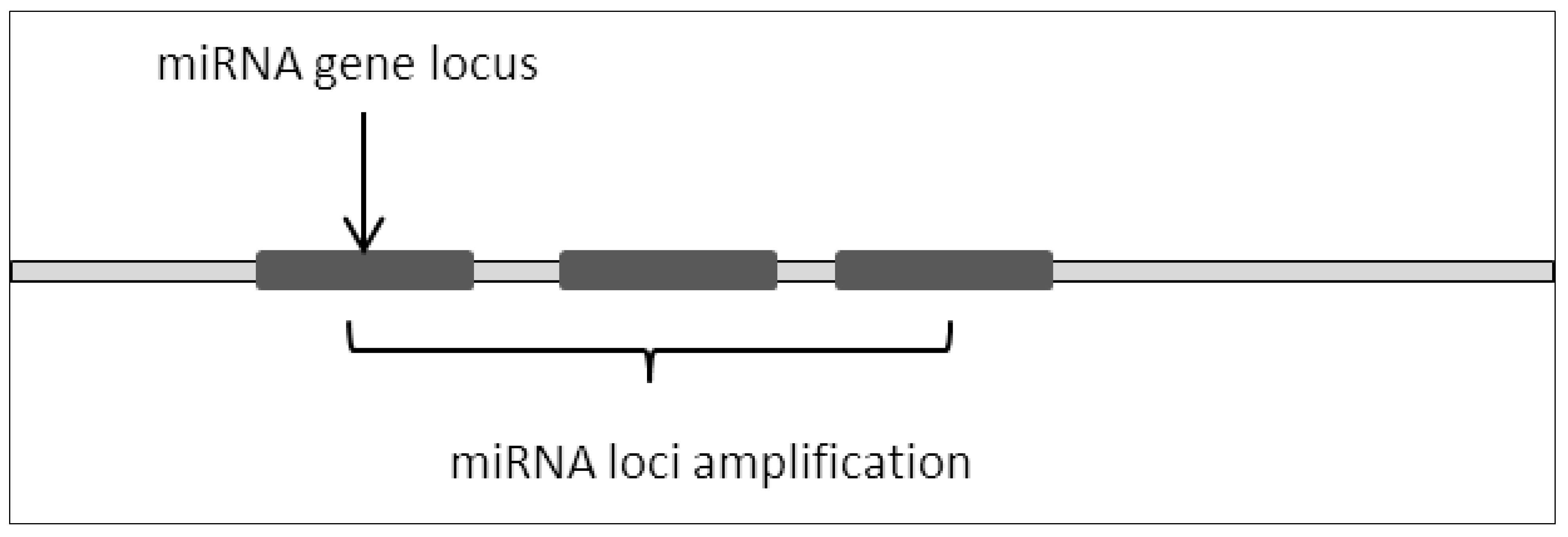
2.1. Amplification of Loci
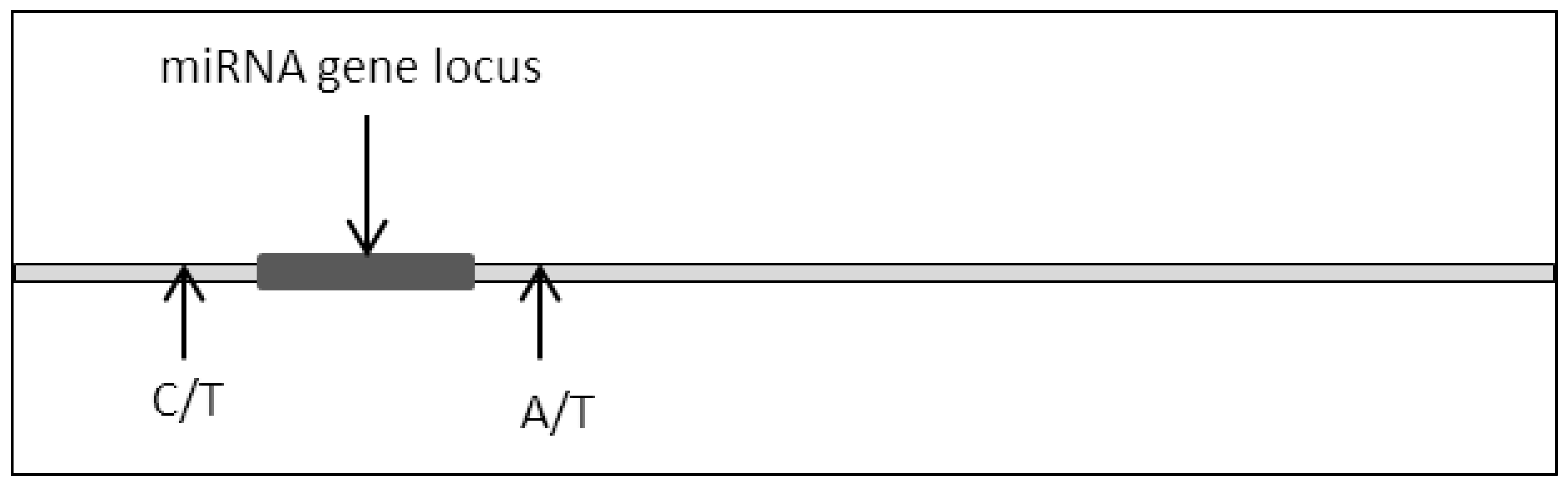
2.2. Mutation (Single Point Mutation, Deletion, Insertion, Base Substitution)
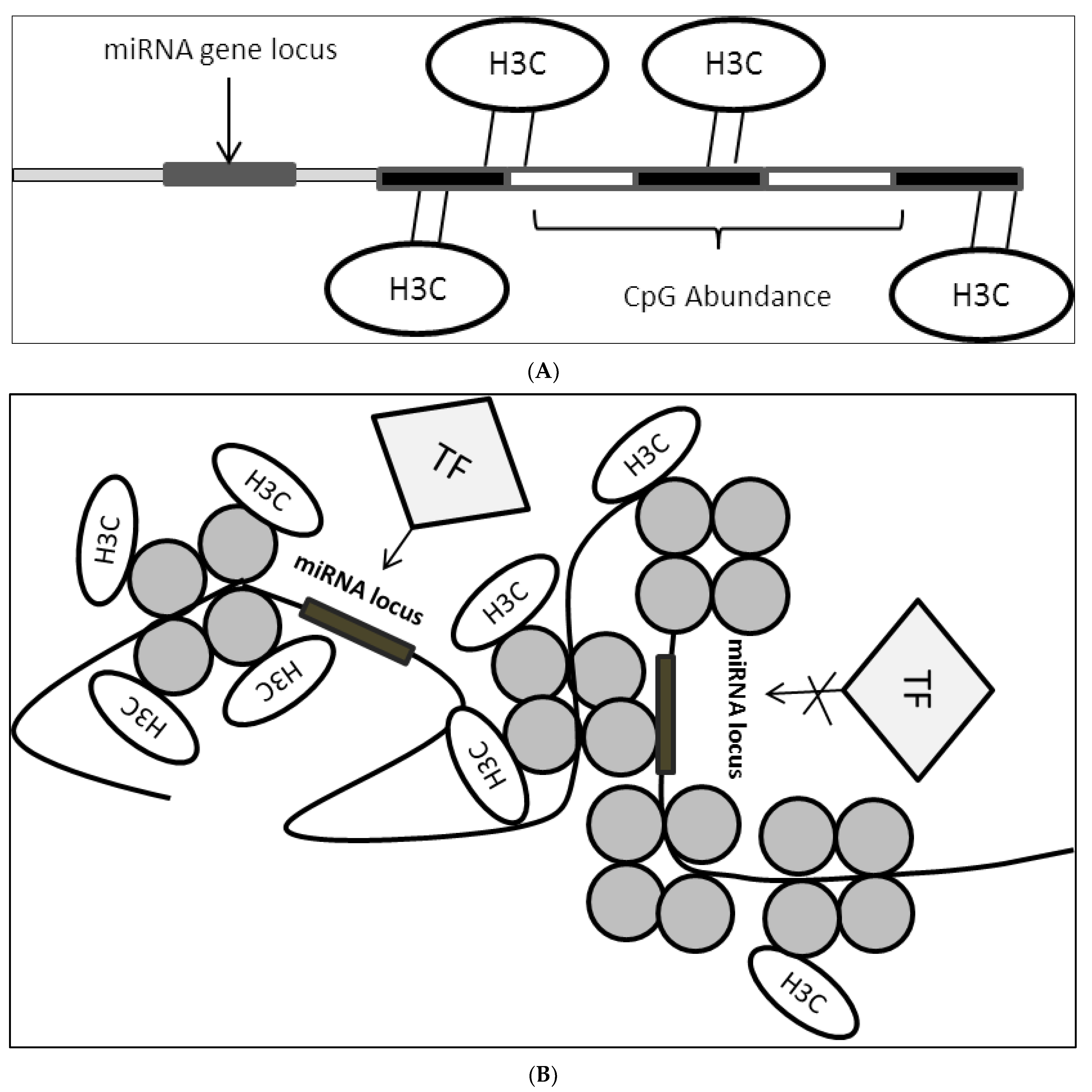
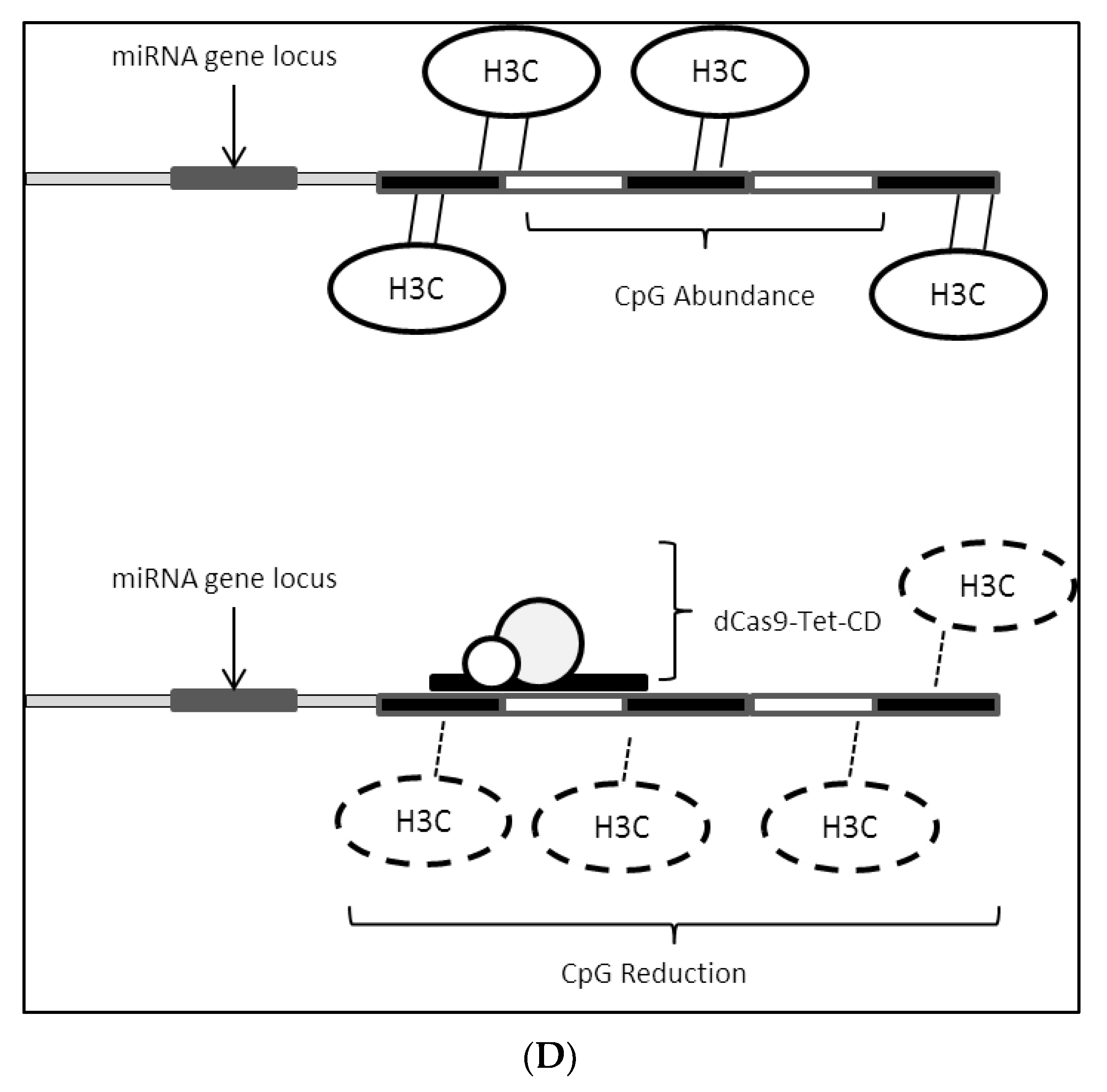
2.3. Epigenetic Modifications on miRNA Loci
2.4. Transcription Factors (TF) Controlling miRNA Expression Dysregulation
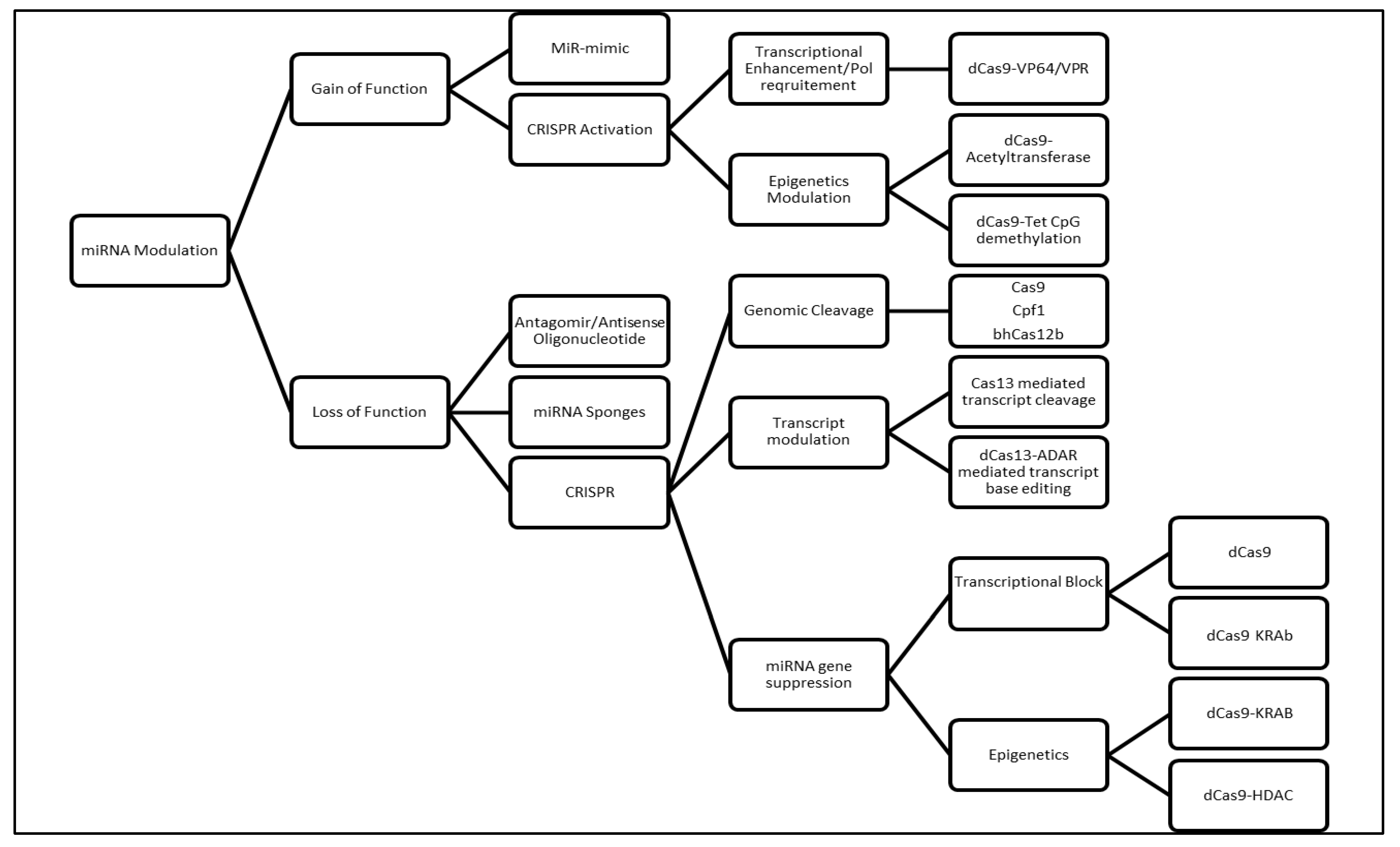
3. Conventional Molecular Genetics Methods for Studying miRNA Dysfunction
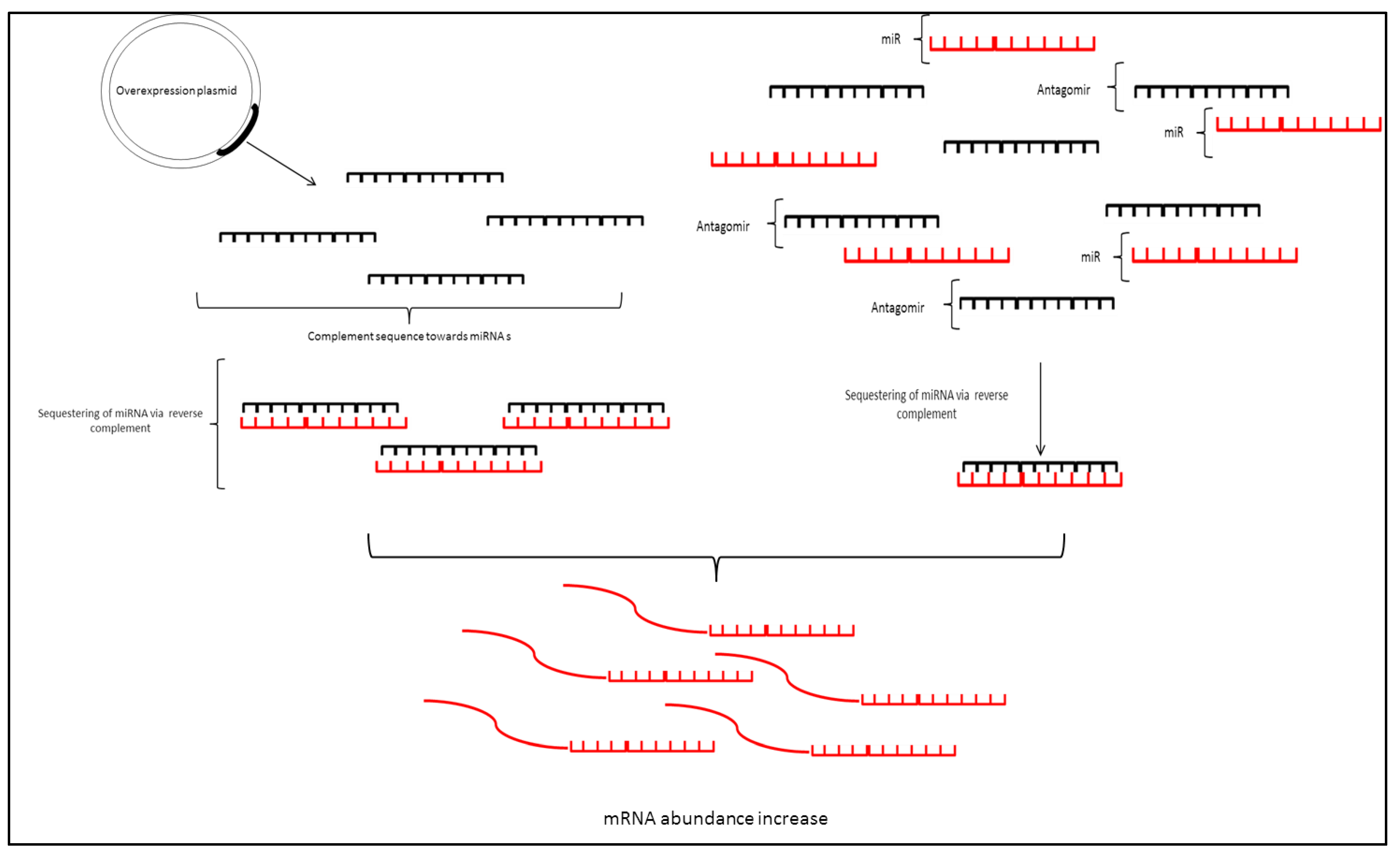
3.1. MicroRNA Sponges
3.2. Antisense Oligonucleotides/Antagomirs
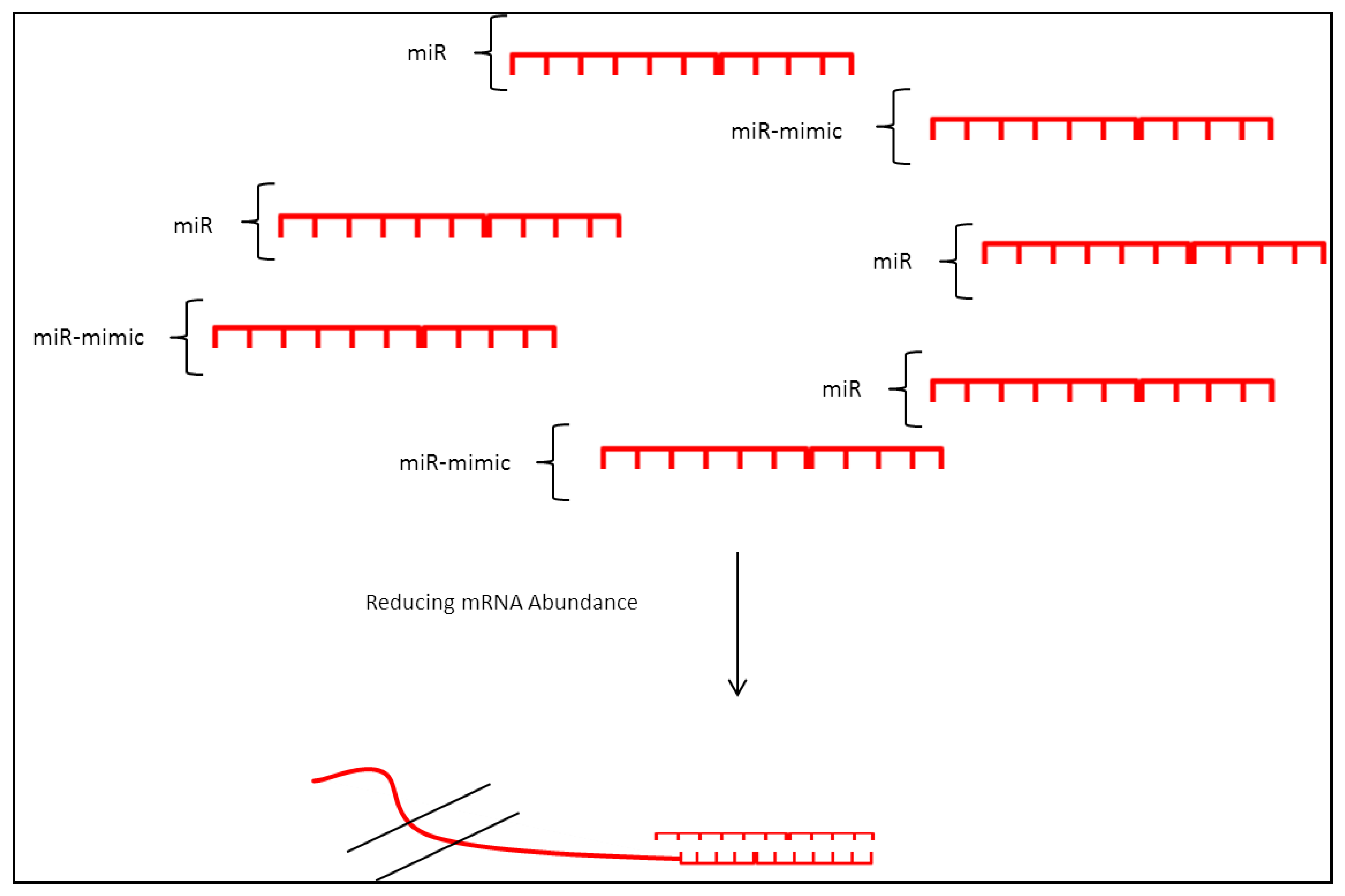
3.3. miRNA Mimic
4. CRISPR/Clustered Regularly Interspaced Short Palindromic Repeats as an Emerging Molecular Genetic Tool to Study miRNA Dysregulation
4.1. CRISPR/Cas9 (CRISPR Associated Protein 9)
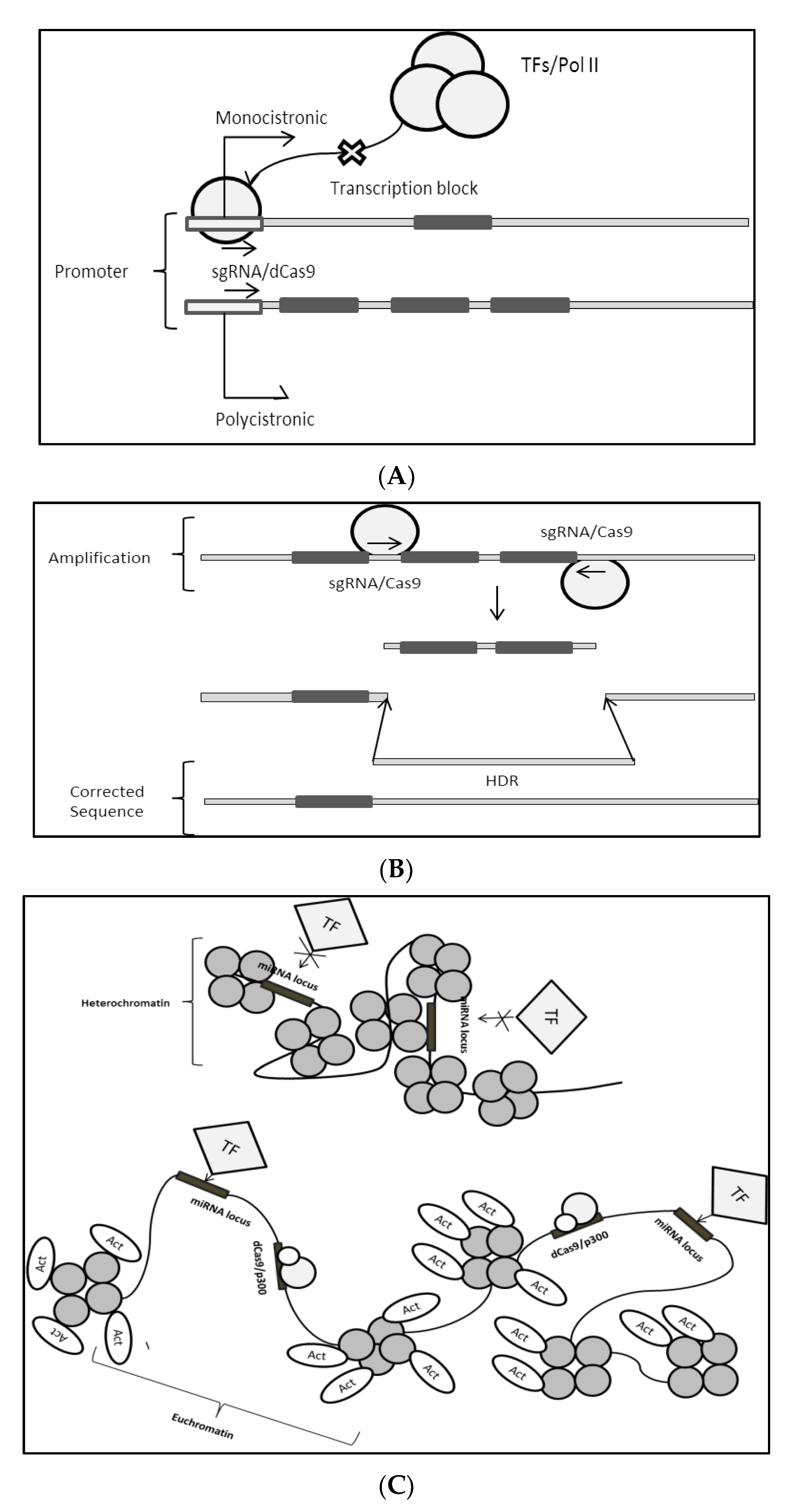
4.1.1. CRISPR/Cas9 Direct Modulation of miRNA Genome
4.1.2. Cas9 Modulation: miRNA Gene Expression Control
4.2. CRISPR/Cpf1 (CRISPR Associated Endonuclease in Prevotella and Francisella 1)
4.3. Cas12b/C2C1
4.4. CRISPR/Cas13
4.5. Utilization of CRISPR to Target miRNA: Practical Considerations
5. Conclusions: Challenges and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Lai, E.C. miRNAs: Whys and wherefores of miRNA-mediated regulation. Curr. Biol. 2005, 15, R458–R460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, E.C. Two decades of miRNA biology: Lessons and challenges. RNA 2015, 21, 675–677. [Google Scholar] [CrossRef]
- Chipman, L.B.; Pasquinelli, A.E. miRNA targeting: Growing beyond the seed. Trends Genet. 2019, 35, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Croce, C.M. miRNA profiling of cancer. Curr. Opin. Genet. Dev. 2013, 23, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Zare, M.; Bastami, M.; Solali, S.; Alivand, M.R. Aberrant miRNA promoter methylation and EMT-involving miRNAs in breast cancer metastasis: Diagnosis and therapeutic implications. J. Cell Physiol. 2018, 233, 3729–3744. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M. microRNAs in cancer susceptibility. Adv. Cancer Res. 2017, 135, 151–171. [Google Scholar]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Chavdoula, E.; Hatziapostolou, M.; Polytarchou, C.; Marcu, K.B.; Papavassiliou, A.G.; Sandaltzopoulos, R.; Kolettas, E. A step-by-step microRNA guide to cancer development and metastasis. Cell. Oncol. 2017, 40, 303–339. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, S.; Jeong, J.M.; Chung, J.-K.; Lee, M.C.; Lee, D.S. Development of a dual-luciferase reporter system for in vivo visualization of MicroRNA biogenesis and posttranscriptional regulation. J. Nucl. Med. 2008, 49, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zhou, Y.; Zhang, Y.; He, L.; Xue, C.; Cao, Y. Design of miRNA sponges for MDV-1 as a therapeutic strategy against lymphomas. Oncotarget 2018, 9, 3842–3852. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Chen, Z.; Liu, X.; Zhou, X. Evaluating the microRNA targeting sites by luciferase reporter gene assay. In MicroRNA Protocols; Springer: Berlin/Heidelberg, Germany, 2013; pp. 117–127. [Google Scholar]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the development of miRNA bioinformatics tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef] [Green Version]
- Ardekani, A.M.; Naeini, M.M. The Role of MicroRNAs in Human Diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Wang, X. Improving microRNA target prediction by modeling with unambiguously identified microRNA-target pairs from CLIP-ligation studies. Bioinformatics 2016, 32, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-H.; Shrestha, S.; Yang, C.-D.; Chang, N.-W.; Lin, Y.-L.; Liao, K.-W.; Huang, W.-C.; Sun, T.-H.; Tu, S.-J.; Lee, W.-H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Chen, Y.-L.; Wang, D.-F.; Wang, Y.-L.; Zhang, T.-P.; Li, H.; Liang, F.; Zhao, Y.; Zhang, G.Y. SgRNA Expression of CRIPSR-Cas9 System Based on MiRNA Polycistrons as a Versatile Tool to Manipulate Multiple and Tissue-Specific Genome Editing. Sci. Rep. 2017, 19, 5795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Liu, D.Z.; Jickling, G.C.; Sharp, F.R.; Yin, K.-J. MicroRNA-based therapeutics in central nervous system injuries. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2018, 38, 1125–1148. [Google Scholar] [CrossRef]
- Yi, B.; Larter, K.; Xi, Y. CRISPR/Cas9 System to Knockdown MicroRNA In Vitro and In Vivo. In Small Non-Coding RNAs: Methods and Protocols; Rederstorff, M., Ed.; Methods in Molecular Biology; Springer US: New York, NY, USA, 2021; pp. 133–139. ISBN 978-1-07-161386-3. [Google Scholar]
- Lataniotis, L.; Albrecht, A.; Kok, F.O.; Monfries, C.A.; Benedetti, L.; Lawson, N.D.; Hughes, S.M.; Steinhofel, K.; Mayr, M.; Zampetaki, A. CRISPR/Cas9 editing reveals novel mechanisms of clustered microRNA regulation and function. Sci. Rep. 2017, 7, 1–14. [Google Scholar]
- Zhou, J.; Deng, K.; Cheng, Y.; Zhong, Z.; Tian, L.; Tang, X.; Tang, A.; Zheng, X.; Zhang, T.; Qi, Y. CRISPR-Cas9 based genome editing reveals new insights into microRNA function and regulation in rice. Front. Plant Sci. 2017, 8, 1598. [Google Scholar] [CrossRef] [Green Version]
- Chaluvally-Raghavan, P.; Jeong, K.J.; Pradeep, S.; Silva, A.M.; Yu, S.; Liu, W.; Moss, T.; Rodriguez-Aguayo, C.; Zhang, D.; Ram, P.; et al. Direct upregulation of STAT3 by microRNA-551b-3p deregulates growth and metastasis of ovarian cancer. Cell Rep. 2016, 15, 1493–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Lee, K.F.; Lu, Y.; Clarke, I.; Shih, D.; Eberhart, C.; Collins, V.P.; Van Meter, T.; Picard, D.; Zhou, L.; et al. Frequent amplification of a chr19q13.41 microRNA polycistron (C19MC) in aggressive primitive neuro-ectodermal brain tumors. Cancer Cell 2009, 16, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, T.; Sin-Chan, P.; Picard, D.; Barszczyk, M.; Hoss, K.; Lu, M.; Kim, S.-K.; Ra, Y.-S.; Nakamura, H.; Fangusaro, J.; et al. CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol. 2014, 128, 291–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A Polycistronic MicroRNA Cluster, miR-17-92, Is Overexpressed in Human Lung Cancers and Enhances Cell Proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, S.; Li, Z.; Chen, P.; He, C.; Cao, D.; Elkahloun, A.; Lu, J.; Pelloso, L.A.; Wunderlich, M.; Huang, H.; et al. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 3710–3715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.; Pan, Y.; Yan, Z.; Li, W.; Cui, J.; Yuan, J.; Tian, L.; Xing, R.; Lu, Y. MiR-23a in amplified 19p13.13 loci targets metallothionein 2A and promotes growth in gastric cancer cells. J. Cell. Biochem. 2013, 114, 2160–2169. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, M.; Chong, Q.-Y.; Zhang, W.; Qian, P.; Yan, H.; Qian, W.; Zhang, M.; Lobie, P.E.; Zhu, T. Amplification of hsa-miR-191/425 locus promotes breast cancer proliferation and metastasis by targeting DICER1. Carcinogenesis 2018, 39, 1506–1516. [Google Scholar] [CrossRef]
- O’Donoghue, P.; Prat, L.; Kucklick, M.; Schäfer, J.G.; Riedel, K.; Rinehart, J.; Söll, D.; Heinemann, I.U. Reducing the genetic code induces massive rearrangement of the proteome. Proc. Natl. Acad. Sci. USA 2014, 111, 17206–17211. [Google Scholar] [CrossRef] [Green Version]
- Drummond, D.A.; Wilke, C.O. The evolutionary consequences of erroneous protein synthesis. Nat. Rev. Genet. 2009, 10, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wong, G.K.-S.; Yu, J. Protein Coding. In Protein Coding; John Wiley & Sons, Ltd: Chichester, UK, 2013; ISBN 978-0-470-01617-6. [Google Scholar]
- Elli, F.M.; Ghirardello, S.; Giavoli, C.; Gangi, S.; Dioni, L.; Crippa, M.; Finelli, P.; Bergamaschi, S.; Mosca, F.; Spada, A. A new structural rearrangement associated to Wolfram syndrome in a child with a partial phenotype. Gene 2012, 509, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Cen, Z.; Xie, F.; Ouyang, Z.; Zhang, B.; Zhao, G.; Luo, W. Genetic analysis of SPG4 and SPG3A genes in a cohort of Chinese patients with hereditary spastic paraplegia. J. Neurol. Sci. 2014, 347, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Azada, M.; Hsiang, D.J.; Herman, J.M.; Kain, T.S.; Siwak-Tapp, C.; Casey, C.; He, J.; Ali, S.M.; Klempner, S.J. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J. Thorac. Oncol. 2014, 9, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, A.; Yamamoto, Y.; Katsumi, A.; Okamoto, A.; Tokuda, M.; Inaguma, Y.; Yamamoto, K.; Yanada, M.; Kanie, T.; Tomita, A. Rearrangement of VPS13B, a causative gene of Cohen syndrome, in a case of RUNX1–RUNX1T1 leukemia with t (8; 12; 21). Int. J. Hematol. 2018, 108, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Porkka, K.P.; Ogg, E.-L.; Saramäki, O.R.; Vessella, R.L.; Pukkila, H.; Lähdesmäki, H.; Weerden, W.M.; van Wolf, M.; Kallioniemi, O.P.; Jenster, G.; et al. The miR-15a-miR-16-1 locus is homozygously deleted in a subset of prostate cancers. Genes Chromosomes Cancer 2011, 50, 499–509. [Google Scholar] [CrossRef]
- Trissal, M.C.; Wong, T.N.; Yao, J.-C.; Ramaswamy, R.; Kuo, I.; Baty, J.; Sun, Y.; Jih, G.; Parikh, N.; Berrien-Elliott, M.M.; et al. MIR142 Loss-of-Function Mutations Derepress ASH1L to Increase HOXA Gene Expression and Promote Leukemogenesis. Cancer Res. 2018, 78, 3510–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanek-Trzeciak, M.O.; Galka-Marciniak, P.; Nawrocka, P.M.; Kowal, E.; Szwec, S.; Giefing, M.; Kozlowski, P. Pan-cancer analysis of somatic mutations in miRNA genes. EBioMedicine 2020, 61, 103051. [Google Scholar] [CrossRef] [PubMed]
- Kwanhian, W.; Lenze, D.; Alles, J.; Motsch, N.; Barth, S.; Döll, C.; Imig, J.; Hummel, M.; Tinguely, M.; Trivedi, P.; et al. MicroRNA-142 is mutated in about 20% of diffuse large B-cell lymphoma. Cancer Med. 2012, 1, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Wehbe-Janek, H.; Henson, R.; Smith, H.; Patel, T. Epigenetic regulation of microRNA-370 by interleukin-6 in malignant human cholangiocytes. Oncogene 2008, 27, 378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorio, M.V.; Piovan, C.; Croce, C.M. Interplay between microRNAs and the epigenetic machinery: An intricate network. Biochim. Biophys. Acta BBA-Gene Regul. Mech. 2010, 1799, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Han, C.; Zhang, J.; Lu, D.; Dash, S.; Feitelson, M.; Lim, K.; Wu, T. Epigenetic regulation of miR-122 by PPARγ and hepatitis B virus X protein in hepatocellular carcinoma cells. Hepatology 2013, 58, 1681–1692. [Google Scholar] [CrossRef] [Green Version]
- Bourassa, M.W.; Ratan, R.R. The interplay between microRNAs and histone deacetylases in neurological diseases. Neurochem. Int. 2014, 77, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasetti, M.; Gaetani, S.; Monaco, F.; Neuzil, J.; Santarelli, L. Epigenetic Regulation of miRNA Expression in Malignant Mesothelioma: miRNAs as Biomarkers of Early Diagnosis and Therapy. Front. Oncol. 2019, 9, 1293. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 1004. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-H.; Hsu, C.-Y.; Tsai, C.-F.; Long, C.-Y.; Wu, C.-H.; Wu, D.-C.; Lee, J.-N.; Chang, W.-C.; Tsai, E.-M. HDAC Inhibitors Target HDAC5, Upregulate MicroRNA-125a-5p, and Induce Apoptosis in Breast Cancer Cells. Mol. Ther. 2015, 23, 656–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.H.; To, K.; Tong, J.H.; Xiao, Z.; Xia, T.; Lai, P.B.S.; Chow, S.C.; Zhu, Y.; Chan, S.L.; Marquez, V.E.; et al. Enhancer of Zeste Homolog 2 Silences MicroRNA-218 in Human Pancreatic Ductal Adenocarcinoma Cells by Inducing Formation of Heterochromatin. Gastroenterology 2013, 144, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Dohi, O.; Yasui, K.; Gen, Y.; Takada, H.; Endo, M.; Tsuji, K.; Konishi, C.; Yamada, N.; Mitsuyoshi, H.; Yagi, N. Epigenetic silencing of miR-335 and its host gene MEST in hepatocellular carcinoma. Int. J. Oncol. 2013, 42, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Goldberger, H.; Dimtchev, A.; Ramalinga, M.; Chijioke, J.; Marian, C.; Oermann, E.K.; Uhm, S.; Kim, J.S.; Chen, L.N. MicroRNA profiling in prostate cancer-the diagnostic potential of urinary miR-205 and miR-214. PLoS ONE 2013, 8, e76994. [Google Scholar] [CrossRef] [Green Version]
- Gandellini, P.; Giannoni, E.; Casamichele, A.; Taddei, M.L.; Callari, M.; Piovan, C.; Valdagni, R.; Pierotti, M.A.; Zaffaroni, N.; Chiarugi, P. miR-205 hinders the malignant interplay between prostate cancer cells and associated fibroblasts. Antioxid. Redox Signal 2014, 20, 1045–1059. [Google Scholar] [CrossRef] [Green Version]
- Hulf, T.; Sibbritt, T.; Wiklund, E.D.; Patterson, K.; Song, J.Z.; Stirzaker, C.; Qu, W.; Nair, S.; Horvath, L.G.; Armstrong, N.J.; et al. Epigenetic-induced repression of microRNA-205 is associated with MED1 activation and a poorer prognosis in localized prostate cancer. Oncogene 2013, 32, 2891–2899. [Google Scholar] [CrossRef] [Green Version]
- Babu, A.; Verma, R.S. Chromosome structure: Euchromatin and heterochromatin. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 1987; Volume 108, pp. 1–60. [Google Scholar]
- Wang, X.; Wang, J.; Ma, H.; Zhang, J.; Zhou, X. Downregulation of miR-195 correlates with lymph node metastasis and poor prognosis in colorectal cancer. Med. Oncol. 2012, 29, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, L.; Xu, Y.; Li, R.; Du, X. microRNA-195 promotes apoptosis and suppresses tumorigenicity of human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2010, 400, 236–240. [Google Scholar] [CrossRef]
- Menigatti, M.; Staiano, T.; Manser, C.N.; Bauerfeind, P.; Komljenovic, A.; Robinson, M.; Jiricny, J.; Buffoli, F.; Marra, G. Epigenetic silencing of monoallelically methylated miRNA loci in precancerous colorectal lesions. Oncogenesis 2013, 2, e56. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Fernandez, R.; Watters, K.; Piskareva, O.; Stallings, R.L.; Bray, I. The role of genetic and epigenetic alterations in neuroblastoma disease pathogenesis. Pediatr. Surg. Int. 2013, 29, 101–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ram Kumar, R.M.; Schor, N.F. Methylation of DNA and chromatin as a mechanism of oncogenesis and therapeutic target in neuroblastoma. Oncotarget 2018, 9, 22184–22193. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Bryan, K.; Buckley, P.G.; Piskareva, O.; Bray, I.M.; Foley, N.; Ryan, J.; Lynch, J.; Creevey, L.; Fay, J.; et al. Modulation of neuroblastoma disease pathogenesis by an extensive network of epigenetically regulated microRNAs. Oncogene 2013, 32, 2927–2936. [Google Scholar] [CrossRef] [Green Version]
- Harbison, C.T.; Gordon, D.B.; Lee, T.I.; Rinaldi, N.J.; Macisaac, K.D.; Danford, T.W.; Hannett, N.M.; Tagne, J.-B.; Reynolds, D.B.; Yoo, J. Transcriptional regulatory code of a eukaryotic genome. Nature 2004, 431, 99. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E.; Dietze, P.; Karas, H.; Knüppel, R. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996, 24, 238–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cawley, S.; Bekiranov, S.; Ng, H.H.; Kapranov, P.; Sekinger, E.A.; Kampa, D.; Piccolboni, A.; Sementchenko, V.; Cheng, J.; Williams, A.J. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 2004, 116, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; He, X.; Li, S.; Sun, R.; Xiang, Q.; Ren, G.; Xiang, T. KRAB zinc-finger protein 382 regulates epithelial-mesenchymal transition and functions as a tumor suppressor, but is silenced by CpG methylation in gastric cancer. Int. J. Oncol. 2018, 53, 961–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488. [Google Scholar] [CrossRef]
- Shu, X.-Z.; Zhang, L.-N.; Zhang, R.; Zhang, C.-J.; He, H.-P.; Zhou, H.; Wang, N.; Zhang, T.-C. Histone acetyltransferase p300 promotes MRTF-A-mediates transactivation of VE-cadherin gene in human umbilical vein endothelial cells. Gene 2015, 563, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Quinn, S.R.; Mangan, N.E.; Caffrey, B.E.; Gantier, M.P.; Williams, B.R.; Hertzog, P.J.; McCoy, C.E.; O’Neill, L.A. The role of Ets2 transcription factor in the induction of microRNA-155 (miR-155) by lipopolysaccharide and its targeting by interleukin-10. J. Biol. Chem. 2014, 289, 4316–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, B.; Lin, J.; Zhao, L. microRNA-29b: An emerging player in human cancer. Asian Pac. J. Cancer Prev. 2014, 15, 9059–9064. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Cai, H.-X.; Gao, S.; Yang, G.-L.; Deng, H.-T.; Xu, G.-C.; Han, J.; Zhang, Q.-Z.; Li, L.-Y. TNFSF15 suppresses VEGF production in endothelial cells by stimulating miR-29b expression via activation of JNK-GATA3 signals. Oncotarget 2016, 7, 69436–69449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-H.; Zhang, W.-J.; Wu, X.-C.; Zhang, M.-D.; Weng, M.-Z.; Zhou, D.; Wang, J.-D.; Quan, Z.-W. Long non-coding RNA Malat1 promotes gallbladder cancer development by acting as a molecular sponge to regulate miR-206. Oncotarget 2016, 7, 37857. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Shen, J.K.; Hornicek, F.J.; Kan, Q.; Duan, Z. Regulation of microRNA-1 (miR-1) expression in human cancer. Biochim. Biophys. Acta BBA-Gene Regul. Mech. 2017, 1860, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Lango-Chavarría, M.; Chimal-Ramírez, G.K.; Ruiz-Tachiquín, M.E.; Espinoza-Sánchez, N.A.; Suárez-Arriaga, M.C.; Fuentes-Pananá, E.M. A 22q11.2 amplification in the region encoding microRNA-650 correlates with the epithelial to mesenchymal transition in breast cancer primary cultures of Mexican patients. Int. J. Oncol. 2017, 50, 432–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, Y.; Murai, N.; Yanaihara, N.; Saito, M.; Saito, M.; Urashima, M.; Murakami, Y.; Matsufuji, S.; Okamoto, A. MicroRNA-21 is a candidate driver gene for 17q23-25 amplification in ovarian clear cell carcinoma. BMC Cancer 2014, 14, 799. [Google Scholar] [CrossRef] [Green Version]
- Bhagirath, D.; Yang, T.L.; Tabatabai, Z.L.; Shahryari, V.; Majid, S.; Dahiya, R.; Tanaka, Y.; Saini, S. Role of a novel race-related tumor suppressor microRNA located in frequently deleted chromosomal locus 8p21 in prostate cancer progression. Carcinogenesis 2019, 40, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Pan, Y.; Bai, L.; Chen, H.; Duan, Z.; Si, Q.; Zhu, R.; Chuang, T.-H.; Luo, Y. MicroRNA-3613-3p functions as a tumor suppressor and represents a novel therapeutic target in breast cancer. Breast Cancer Res. 2021, 23, 12. [Google Scholar] [CrossRef]
- Galka-Marciniak, P.; Urbanek-Trzeciak, M.O.; Nawrocka, P.M.; Dutkiewicz, A.; Giefing, M.; Lewandowska, M.A.; Kozlowski, P. Somatic Mutations in miRNA Genes in Lung Cancer—Potential Functional Consequences of Non-Coding Sequence Variants. Cancers 2019, 11, 793. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; He, Q.; Wen, X.; Hong, X.; Yang, X.; Tang, X.; Zhang, P.; Lei, Y.; Sun, Y.; Zhang, J.; et al. EZH2-DNMT1-mediated epigenetic silencing of miR-142-3p promotes metastasis through targeting ZEB2 in nasopharyngeal carcinoma. Cell Death Differ. 2019, 26, 1089–1106. [Google Scholar] [CrossRef]
- Bhatia, V.; Yadav, A.; Tiwari, R.; Nigam, S.; Goel, S.; Carskadon, S.; Gupta, N.; Goel, A.; Palanisamy, N.; Ateeq, B. Epigenetic Silencing of miRNA-338-5p and miRNA-421 Drives SPINK1-Positive Prostate Cancer. Clin. Cancer Res. 2019, 25, 2755–2768. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-G.; Shi, Q.-W.; Yuan, L.; Qin, L.; Wang, Y.; Miao, Y.-Q.; Chen, Z.; Ling, C.-Q.; Qin, W. C-Myc-dependent repression of two oncogenic miRNA clusters contributes to triptolide-induced cell death in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 51. [Google Scholar] [CrossRef]
- Wang, B.; Hsu, S.; Wang, X.; Kutay, H.; Bid, H.K.; Yu, J.; Ganju, R.K.; Jacob, S.T.; Yuneva, M.; Ghoshal, K. Reciprocal regulation of microRNA-122 and c-Myc in hepatocellular cancer: Role of E2F1 and transcription factor dimerization partner 2. Hepatology 2014, 59, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La, T.; Liu, G.Z.; Farrelly, M.; Cole, N.; Feng, Y.C.; Zhang, Y.Y.; Sherwin, S.K.; Yari, H.; Tabatabaee, H.; Yan, X.G.; et al. A p53-Responsive miRNA Network Promotes Cancer Cell Quiescence. Cancer Res. 2018, 78, 6666–6679. [Google Scholar] [CrossRef] [Green Version]
- Bak, R.O.; Mikkelsen, J.G. miRNA sponges: Soaking up miRNAs for regulation of gene expression. Wiley Interdiscip. Rev. RNA 2014, 5, 317–333. [Google Scholar] [CrossRef]
- Wang, P.; Zhi, H.; Zhang, Y.; Liu, Y.; Zhang, J.; Gao, Y.; Guo, M.; Ning, S.; Li, X. miRSponge: A manually curated database for experimentally supported miRNA sponges and ceRNAs. Database 2015, 2015, bav098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, S.; Wu, J.; Cui, J.; Zhong, L.; Zeng, L.; Ge, S. circRNA_100290 plays a role in oral cancer by functioning as a sponge of the miR-29 family. Oncogene 2017, 36, 4551. [Google Scholar] [CrossRef]
- Van Peer, G.; Mets, E.; Claeys, S.; De Punt, I.; Lefever, S.; Ongenaert, M.; Rondou, P.; Speleman, F.; Mestdagh, P.; Vandesompele, J. A high-throughput 3’UTR reporter screening identifies microRNA interactomes of cancer genes. PLoS ONE 2018, 13, e0194017. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Li, C.; Chen, J.; Zhang, K.; Chu, X.; Wang, R.; Chen, L. The emerging roles of long noncoding RNA ROR (lincRNA-ROR) and its possible mechanisms in human cancers. Cell. Physiol. Biochem. 2016, 40, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Gao, Q.; Wang, J.; Zhang, X.; Liu, K.; Duan, Z. Linc-RNA-RoR acts as a “sponge” against mediation of the differentiation of endometrial cancer stem cells by microRNA-145. Gynecol. Oncol. 2014, 133, 333–339. [Google Scholar] [CrossRef]
- Bofill-De Ros, X.; Santos, M.; Vila-Casadesús, M.; Villanueva, E.; Andreu, N.; Dierssen, M.; Fillat, C. Genome-wide miR-155 and miR-802 target gene identification in the hippocampus of Ts65Dn Down syndrome mouse model by miRNA sponges. BMC Genom. 2015, 16, 907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Cheng, Y.; Li, P.; Tao, J.; Deng, X.; Zhang, X.; Gu, M.; Lu, Q.; Yin, C. A lentiviral sponge for miRNA-21 diminishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumor Biol. 2015, 36, 383–391. [Google Scholar] [CrossRef]
- Wang, X. Composition of seed sequence is a major determinant of microRNA targeting patterns. Bioinformatics 2014, 30, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [Green Version]
- Ebert, M.S.; Sharp, P.A. Emerging Roles for Natural MicroRNA Sponges. Curr. Biol. 2010, 20, R858–R861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, N.; Stein, C.A. Antisense Oligonucleotides: Basic Concepts and Mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [PubMed]
- Zhou, S.; Wang, Y.; Meng, Y.; Xiao, C.; Liu, Z.; Brohawn, P.; Higgs, B.W.; Jallal, B.; Jia, Q.; Qu, B. In Vivo Therapeutic Success of MicroRNA-155 Antagomir in a Mouse Model of Lupus Alveolar Hemorrhage. Arthritis Rheumatol. 2016, 68, 953–964. [Google Scholar] [CrossRef]
- Dorrance, A.M.; Neviani, P.; Ferenchak, G.J.; Huang, X.; Nicolet, D.; Maharry, K.S.; Ozer, H.G.; Hoellarbauer, P.; Khalife, J.; Hill, E.B. Targeting leukemia stem cells in vivo with antagomiR-126 nanoparticles in acute myeloid leukemia. Leukemia 2015, 29, 2143. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.; Vuppusetty, C.; Ito, K.; Barnes, P.J. Antagomir Of Microrna-34a Rescues Cellular Senescence In Bronchial Epithelial Cells Of COPD Patients. In D108 Cell Fate Decisions: Senescence, Repair, and Regeneration; American Thoracic Society: Oxford, UK, 2017; p. A7490. [Google Scholar]
- Yu, C.; Qian, L.; Uttamchandani, M.; Li, L.; Yao, S.Q. Single-vehicular delivery of antagomir and small molecules to inhibit miR-122 function in hepatocellular carcinoma cells by using “smart” mesoporous silica nanoparticles. Angew. Chem. Int. Ed. 2015, 54, 10574–10578. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-J.; Ouyang, Y.-B.; Xiong, X.; Stary, C.M.; Giffard, R.G. Post-stroke treatment with miR-181 antagomir reduces injury and improves long-term behavioral recovery in mice after focal cerebral ischemia. Exp. Neurol. 2015, 264, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Brunschweiger, A.; Gebert, L.F.; Lucic, M.; Pradère, U.; Jahns, H.; Berk, C.; Hunziker, J.; Hall, J. Site-specific conjugation of drug-like fragments to an antimiR scaffold as a strategy to target miRNAs inside RISC. Chem. Commun. 2016, 52, 156–159. [Google Scholar] [CrossRef] [Green Version]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting noncoding RNAs in disease. J. Clin. Investig. 2017, 127, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.-Y.; Xiao, W.-Z.; Wang, F.; Wang, Y.-Q.; Zhu, Y.-H.; Wu, Y.-F.; Miao, Z.-L.; Lin, Y.-C. MicroRNA-320 inhibits cell proliferation in glioma by targeting E2F1. Mol. Med. Rep. 2015, 12, 2355–2359. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Wang, K.; Wang, G.; Zhang, A.; Pu, P. MiR-30a-5p Antisense Oligonucleotide Suppresses Glioma Cell Growth by Targeting SEPT7. PLoS ONE 2013, 8, e55008. [Google Scholar] [CrossRef]
- Li, Z.; Chen, P.; Su, R.; Li, Y.; Hu, C.; Wang, Y.; Arnovitz, S.; He, M.; Gurbuxani, S.; Zuo, Z. Overexpression and knockout of miR-126 both promote leukemogenesis through targeting distinct gene signaling. Blood 2015, 126, 3667. [Google Scholar]
- Brewer, C.J.; Cook, G.J.; Zhang, B.; Qi, J.; Hua, W.-K.; Cai, Q.; Carnahan, E.; Marom, A.; Li, L.; Kortylewski, M. MiR-126 Promotes Leukemogenesis in Inv (16) Acute Myeloid Leukemia. Am. Soc. Hematol. 2017, 130, 301. [Google Scholar]
- Sharma, V.K.; Sharma, R.K.; Singh, S.K. Antisense oligonucleotides: Modifications and clinical trials. MedChemComm 2014, 5, 1454–1471. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222. [Google Scholar] [CrossRef] [PubMed]
- Braasch, D.A.; Corey, D.R. Locked nucleic acid (LNA): Fine-tuning the recognition of DNA and RNA. Chem. Biol. 2001, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kurreck, J. Antisense technologies: Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef]
- Deleavey, G.F.; Damha, M.J. Designing Chemically Modified Oligonucleotides for Targeted Gene Silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef] [Green Version]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef]
- Reid, G.; Kao, S.C.; Pavlakis, N.; Brahmbhatt, H.; MacDiarmid, J.; Clarke, S.; Boyer, M.; van Zandwijk, N. Clinical development of TargomiRs, a miRNA mimic-based treatment for patients with recurrent thoracic cancer. Epigenomics 2016, 8, 1079–1085. [Google Scholar] [CrossRef] [Green Version]
- Cutrona, G.; Matis, S.; Colombo, M.; Massucco, C.; Baio, G.; Valdora, F.; Emionite, L.; Fabris, S.; Recchia, A.G.; Gentile, M.; et al. Effects of miRNA-15 and miRNA-16 expression replacement in chronic lymphocytic leukemia: Implication for therapy. Leukemia 2017, 31, 1894–1904. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.; Kim, W.K.; Kwon, Y.; Jang, M.; Bauer, S.; Kim, H. Survivin is a novel transcription regulator of KIT and is downregulated by miRNA-494 in gastrointestinal stromal tumors. Int. J. Cancer 2018, 142, 2080–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibayama, Y.; Kondo, T.; Ohya, H.; Fujisawa, S.-I.; Teshima, T.; Iseki, K. Upregulation of microRNA-126-5p is associated with drug resistance to cytarabine and poor prognosis in AML patients. Oncol. Rep. 2015, 33, 2176–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Søkilde, R.; Newie, I.; Persson, H.; Borg, A.; Rovira, C. Passenger strand loading in overexpression experiments using microRNA mimics. RNA Biol. 2015, 12, 787–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.Y.; Gonzalez-Martin, A.; Miletic, A.V.; Lai, M.; Knight, S.; Sabouri-Ghomi, M.; Head, S.R.; Macauley, M.S.; Rickert, R.C.; Xiao, C. Transfection of microRNA Mimics Should Be Used with Caution. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Abraham, J.M.; Cheng, Y.; Wang, Z.; Wang, Z.; Zhang, G.; Ashktorab, H.; Smoot, D.T.; Cole, R.N.; Boronina, T.N.; et al. Synthetic Circular RNA Functions as a miR-21 Sponge to Suppress Gastric Carcinoma Cell Proliferation. Mol. Ther. Nucleic Acids 2018, 13, 312–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Ma, K.; Cheng, Y.; Abraham, J.M.; Liu, X.; Ke, X.; Wang, Z.; Meltzer, S.J. Synthetic circular multi-miR sponge simultaneously inhibits miR-21 and miR-93 in esophageal carcinoma. Lab. Investig. 2019, 99, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Wu, K.; Zeng, Z.; Huang, S.; Ji, X.; Yuan, C.; Zhang, L.; Liu, W.; Huang, B.; Feng, Y.; et al. A Simplified System to Express Circularized Inhibitors of miRNA for Stable and Potent Suppression of miRNA Functions. Mol. Ther. Nucleic Acids 2018, 13, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Song, C.; Wang, X.; Li, Y.; Bai, X.; Liang, X.; Wu, J.; Liu, J. Downregulation of miR-155-5p enhances the anti-tumor effect of cetuximab on triple-negative breast cancer cells via inducing cell apoptosis and pyroptosis. Aging 2021, 13, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hu, Z.; Wang, Z.; Zhu, T.; Wang, G.; Gao, B.; Wang, J.; Deng, X. miR-125a-5p promotes gastric cancer growth and invasion by regulating the Hippo pathway. J. Clin. Lab. Anal. 2021, 28, e24078. [Google Scholar] [CrossRef]
- Kardani, A.; Yaghoobi, H.; Alibakhshi, A.; Khatami, M. Inhibition of miR-155 in MCF-7 breast cancer cell line by gold nanoparticles functionalized with antagomir and AS1411 aptamer. J. Cell. Physiol. 2020, 235, 6887–6895. [Google Scholar] [CrossRef]
- Ueda, S.; Takanashi, M.; Sudo, K.; Kanekura, K.; Kuroda, M. miR-27a ameliorates chemoresistance of breast cancer cells by disruption of reactive oxygen species homeostasis and impairment of autophagy. Lab. Investig. 2020, 100, 863–873. [Google Scholar] [CrossRef]
- Fang, Z.H.; Wang, S.L.; Zhao, J.T.; Lin, Z.J.; Chen, L.Y.; Su, R.; Xie, S.T.; Carter, B.Z.; Xu, B. miR-150 exerts antileukemia activity in vitro and in vivo through regulating genes in multiple pathways. Cell Death Dis. 2016, 7, e2371. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Lee, A.S.Y.; Engelman, A.; Doudna, J.A. Integrase-mediated spacer acquisition during CRISPR–Cas adaptive immunity. Nature 2015, 519, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marraffini, L.A. CRISPR-Cas immunity in prokaryotes. Nature 2015, 526, 55. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Santa-Inez, D.C.; Fuziwara, C.S.; Saito, K.C.; Kimura, E.T. Targeting the Highly Expressed microRNA miR-146b with CRISPR/Cas9n Gene Editing System in Thyroid Cancer. Int. J. Mol. Sci. 2021, 22, 7992. [Google Scholar] [CrossRef]
- Jiang, F.-N.; Liang, Y.-X.; Wei, W.; Zou, C.-Y.; Chen, G.-X.; Wan, Y.-P.; Liu, Z.-Z.; Yang, Y.; Han, Z.-D.; Zhu, J.-G.; et al. Functional classification of prostate cancer-associated miRNAs through CRISPR/Cas9-mediated gene knockout. Mol. Med. Rep. 2020, 22, 3777–3784. [Google Scholar] [CrossRef]
- Chang, H.; Yi, B.; Ma, R.; Zhang, X.; Zhao, H.; Xi, Y. CRISPR/cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci. Rep. 2016, 6, 22312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, H.; Yonemori, M.; Miyamoto, K.; Tatarano, S.; Kofuji, S.; Nohata, N.; Nakagawa, M.; Enokida, H. microRNA-210-3p depletion by CRISPR/Cas9 promoted tumorigenesis through revival of TWIST1 in renal cell carcinoma. Oncotarget 2017, 8, 20881–20894. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Kanamori, M.; Someya, K.; Nakatsukasa, H.; Yoshimura, A. Stabilization of Foxp3 expression by CRISPR-dCas9-based epigenome editing in mouse primary T cells. Epigenetics Chromatin 2017, 10, 24. [Google Scholar] [CrossRef]
- Wang, H.; Guo, R.; Du, Z.; Bai, L.; Li, L.; Cui, J.; Li, W.; Hoffman, A.R.; Hu, J.-F. Epigenetic targeting of Granulin in hepatoma cells by synthetic CRISPR dCas9 epi-suppressors. Mol. Ther. Nucleic Acids 2018, 11, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9–based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [Green Version]
- Polstein, L.R.; Gersbach, C.A. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 2015, 11, 198–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the epigenome: Technologies for programmable transcription and epigenetic modulation. Nat. Methods 2016, 13, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Ji, H.; Kabadi, A.M.; Gersbach, C.A.; Christoforou, N.; Leong, K.W. A CRISPR/Cas9-Based System for Reprogramming Cell Lineage Specification. Stem Cell Rep. 2014, 3, 940–947. [Google Scholar] [CrossRef] [Green Version]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddalo, D.; Manchado, E.; Concepcion, C.P.; Bonetti, C.; Vidigal, J.A.; Han, Y.-C.; Ogrodowski, P.; Crippa, A.; Rekhtman, N.; de Stanchina, E.; et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 2014, 516, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, T.; Nishikawa, A.; Kume, S.; Chayama, K.; Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014, 4, 5400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordin, M.; D’Atri, F.; Guillemot, L.; Citi, S. Histone Deacetylase Inhibitors Up-Regulate the Expression of Tight Junction Proteins11Swiss Cancer League, Swiss National Science Foundation, Ministry for Italian University and Research, ERASMUS Program (M. Bordin), and Roche Research Foundation fellowship (L. Guillemot). Mol. Cancer Res. 2004, 2, 692–701. [Google Scholar] [PubMed]
- Yang, S.; Zhang, J.; Zhang, Y.; Wan, X.; Zhang, C.; Huang, X.; Huang, W.; Pu, H.; Pei, C.; Wu, H. KDM1A triggers androgen-induced miRNA transcription via H3K4me2 demethylation and DNA oxidation. Prostate 2015, 75, 936–946. [Google Scholar] [CrossRef]
- Georgakilas, G.; Perdikopanis, N.; Hatzigeorgiou, A.G. Identifying Pri-miRNA Transcription Start Sites. In miRNA Biogenesis; Springer: Berlin/Heidelberg, Germany, 2018; pp. 11–31. [Google Scholar]
- Zhao, Y.; Dai, Z.; Liang, Y.; Yin, M.; Ma, K.; He, M.; Ouyang, H.; Teng, C.-B. Sequence-specific inhibition of microRNA via CRISPR/CRISPRi system. Sci. Rep. 2014, 4, 3943. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresca, M.; Lin, V.G.; Guo, N.; Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): Custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 2013, 23, 539–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, W.; Shan, L.; Han, L.; Ma, S.; Zhang, Y.; Hao, B.; Lin, Y.; Rong, Z. Gene activation in human cells using CRISPR/Cpf1-p300 and CRISPR/Cpf1-SunTag systems. Protein Cell 2018, 9, 380–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Han, J.; Chen, Z.; Wu, H.; Dong, H.; Nie, G. Engineering cell signaling using tunable CRISPR–Cpf1-based transcription factors. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozsolak, F.; Poling, L.L.; Wang, Z.; Liu, H.; Liu, X.S.; Roeder, R.G.; Zhang, X.; Song, J.S.; Fisher, D.E. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008, 22, 3172–3183. [Google Scholar] [CrossRef] [Green Version]
- Strecker, J.; Jones, S.; Koopal, B.; Schmid-Burgk, J.; Zetsche, B.; Gao, L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.; Abudayyeh, O.; Gootenberg, J.; Zhang, F. CRISPR Tools for Systematic Studies of RNA Regulation. Cold Spring Harb. Perspect. Biol. 2019, 11, a035386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR–Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of MicroRNA–Target Recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- Olena, A.F.; Patton, J.G. Genomic organization of microRNAs. J. Cell. Physiol. 2010, 222, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Ratnadiwakara, M.; Mohenska, M.; Änkö, M.-L. Splicing factors as regulators of miRNA biogenesis–links to human disease. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 79, pp. 113–122. [Google Scholar]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.; Peking, P.; Klausegger, A.; Murauer, E.M.; Hofbauer, J.P.; Wally, V.; Lettner, T.; Hainzl, S.; Ablinger, M.; Bauer, J.W.; et al. Cut and Paste: Efficient Homology-Directed Repair of a Dominant Negative KRT14 Mutation via CRISPR/Cas9 Nickases. Mol. Ther. 2017, 25, 2585–2598. [Google Scholar] [CrossRef] [Green Version]
- Zlotorynski, E. Epigenetics: Characterizing enhancers with dCas9. Nat. Rev. Mol. Cell Biol. 2015, 16, 266. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Tang, D.; Leong, K.W. CRISPR/dCas9-mediated cell differentiation. Curr. Opin. Biomed. Eng. 2018, 7, 9–15. [Google Scholar] [CrossRef]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tollefsbol, T.O. DNA methylation detection: Bisulfite genomic sequencing analysis. In Epigenetics Protocols; Springer: Berlin/Heidelberg, Germany, 2011; pp. 11–21. [Google Scholar]
- Jiang, Q.; Meng, X.; Meng, L.; Chang, N.; Xiong, J.; Cao, H.; Liang, Z. Small indels induced by CRISPR/Cas9 in the 5′ region of microRNA lead to its depletion and Drosha processing retardance. RNA Biol. 2014, 11, 1243–1249. [Google Scholar] [CrossRef] [Green Version]
- Huo, W.; Zhao, G.; Yin, J.; Ouyang, X.; Wang, Y.; Yang, C.; Wang, B.; Dong, P.; Wang, Z.; Watari, H. Lentiviral CRISPR/Cas9 vector mediated miR-21 gene editing inhibits the epithelial to mesenchymal transition in ovarian cancer cells. J. Cancer 2017, 8, 57. [Google Scholar] [CrossRef]
- Abdollah, N.A.; Kumitaa, T.D.; Narazah, M.Y.; Razak, S.R.A. Sequence-specific inhibition of microRNA-130a gene by CRISPR/Cas9 system in breast cancer cell line. J. Phys. Conf. Ser. 2017, 851, 012037. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, W.; Zeng, W.; Wan, C.; Duan, S.; Jiang, S. microRNA-137 promotes apoptosis in ovarian cancer cells via the regulation of XIAP. Br. J. Cancer 2017, 116, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Li, Q.; Du, J.; Li, X.; Jiang, S.; He, Y. Establishment of an miR-137-knockout cell model using CRISPR/Cas9 genome editing. Oncol. Lett. 2018, 16, 4027–4032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, S.; Aich, M.; Kumar, A.; Sengupta, S.; Bajad, P.; Dhapola, P.; Paul, D.; Narta, K.; Purkrait, S.; Mehani, B.; et al. Novel internal regulators and candidate miRNAs within miR-379/miR-656 miRNA cluster can alter cellular phenotype of human glioblastoma. Sci. Rep. 2018, 8, 7673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannafon, B.N.; Cai, A.; Calloway, C.L.; Xu, Y.-F.; Zhang, R.; Fung, K.-M.; Ding, W.-Q. miR-23b and miR-27b are oncogenic microRNAs in breast cancer: Evidence from a CRISPR/Cas9 deletion study. BMC Cancer 2019, 19, 642. [Google Scholar] [CrossRef]
- Akdemir, B.; Nakajima, Y.; Inazawa, J.; Inoue, J. miR-432 Induces NRF2 Stabilization by Directly Targeting KEAP1. Mol. Cancer Res. 2017, 15, 1570–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuziwara, C.S.; Saito, K.C.; Kimura, E.T. Thyroid Follicular Cell Loss of Differentiation Induced by MicroRNA miR-17-92 Cluster Is Attenuated by CRISPR/Cas9n Gene Silencing in Anaplastic Thyroid Cancer. Thyroid 2019, 30, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.-L.; Wang, X.-H.; Sun, B.-F.; Zhang, X.-D.; Zhu, X.-H.; Yu, Z.-J.; Luo, H. Expression, regulation and mechanism of action of the miR-17-92 cluster in tumor cells. Int. J. Mol. Med. 2017, 40, 1624–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajic, M.; Froio, D.; Daly, S.; Doculara, L.; Millar, E.; Graham, P.H.; Drury, A.; Steinmann, A.; Bock, C.E.; de Boulghourjian, A.; et al. miR-139-5p Modulates Radiotherapy Resistance in Breast Cancer by Repressing Multiple Gene Networks of DNA Repair and ROS Defense. Cancer Res. 2018, 78, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.P.; Knutsen, E.; Calin, G.A. Classical and noncanonical functions of miRNAs in cancers. Trends Genet. 2021. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Mechanism of Dysregulation | Consequence | References |
|---|---|---|---|
| miR-650 | Loci amplification | Inverse correlation was observed between miR-650 and tumour supressor genes ING4 and NDRG2 | [77] |
| miR-21 | Loci amplification | miR-21 overexpression leads to PTEN suppression | [78] |
| miR-4288 | Deletion | Loss of miRNA in prostate cancer, miRNA directly represses metastatic/invasion genes MMP16 and ROCK1 | [79] |
| miR-3613 | Deletion | miR-3613 was found to be lower in breast cancer. Gain of function reveals miR-3613 to regulate PAFAH1B2 and PDK3 blocking oncogenesis | [80] |
| miR-379 | Base substitution | Hotspot mutation commonly occurring in lung adenocarcinoma | [81] |
| miR-142-3p | Epigenetic suppression via DNMT recruitment | Hypermethylation of miR-142 leads to unfavorable prognosis in nasopharyngeal carcinoma | [82] |
| miR-338-5p/421 | EZH2 mediated suppression via DNA methylation | Presence of CpG marks on primary prostate cancer. Ectopic expression reveals suppression in prostate cancer growth | [83] |
| miR-17-92/106b-25 | CMYC driven | CMYC drive the expression of these miRNA clusters, inhibition of cMYC activators resulted in suppression of these clusters in hepatocellular carcinoma | [84] |
| miR-122 | CMYC driven | CMYC oncogene overexpression in hepatocellular carcinoma activates miR-122 via direct promoter binding driving oncogenesis | [85] |
| miR-455-3p | Reside in host gene driven by p53 | miRNA involves in cancer quiescence via p53 mediation | [86] |
| Method of Study | miRNA | Mechanism | Outcome | References |
|---|---|---|---|---|
| miRNA sponges | miR-21 | Synthetic RNA sponge was designed to bear sequence complementary to miR-21 | Downregulated proteins due to miR-21 overexpression were restored | [124] |
| miR-21 and miR-93 | Single RNA sponges bearing multiple complementary sites against target miRNAs | Targetting oncomiRs effectively induce apoptosis and blocked proliferation of esophageal carcinoma | [125] | |
| miR-223 | RNA sponge Expressing DNA plasmid was used. RNA circularization was imposed using slicing acceptor and donor site | Sponges effectively sequester endogenous miRNA in T-ALL cells effective restoration of miR target genes | [126] | |
| Antagomirs/Antisense Oligo nucleotide | miR-155-5p | Transfection of breast cancer cell lines with antagonist against miR | Downregulation of miR leads to increase in breast cancer sensitivity against cetuximab | [127] |
| miR-125a-5p | Transfection of gastric cancer cells with antagomir | Restoration of miR suppressed genes was observed, suppression leads to the suppression in EMT of gastric cancer | [128] | |
| miR-155 | Delivery of antagomir into MCF-7 via attachment to gold nano particle | Elevation in miR target gene T53INP1 was observed stimulating apoptosis of breast cancer cells | [129] | |
| miRNA mimics | miR-27a | Overexpression of miR via mimics transfection Mimics used are as standard siRNA sizes | Overexpression of miR-27a via mimic alleviates cancer characteristics and sensitizes breast cancer towards anticancer drugs | [130] |
| miR-150 | miRNA sequence was expressed using pEZX-MR via lentiviral delivery | Mimic expression induces apoptosis in multiple leukemic cell lines | [131] |
| microRNA Target | Target Site | CRISPR System, Delivery | Model | Outcome |
|---|---|---|---|---|
| miR-93 [180] | 5′ Drosha Processing site | CRISPR/Cas9, Lipofection | Human Cervical Cancer (HeLa) | Almost no detection of mature miR-93 Accumulation of primary miR-93 transcript suggest impairment in Drosha processing |
| miR-21 [181] | 20nt sequence adjacent to PAM (NGG) | CRISPR/Cas9, Lentiviral Vector | Human Ovarian Adenocarcinoma (SKOV3 and OVCR3) | Significant reduction in mature miR-21 expression was observed |
| miR-130a [182] | 5p and 3p Seed Sequence Stem Loop (Dicer binding Site) | CRISPR/Cas9, Lipofection | Human Breast Cancer (MCF7) | Significant reduction was observed when using Cas9 targeting from the 5p region No significant difference in miR-130a expression was observed when targeting either the 3p or the Stem Loop sequence |
| miR-137 [183,184] | Nucleotide sequence upstream of 5′ PAM (NGG) | CRISPR/Cas9, Lentiviral Vector | Human Ovarian Carcinoma (A2780) | Significant reduction in mature miR-137 expression was observed. Deletion and insertion mutation detected from single-cell expanded colonies. |
| miR-379/miR-656 cluster [185] | dCas9 fused to VP-64 docking on the miRNA locus for induction of miRNA gene expression. | CRISPR/dCas9, Lipofection | Human Glioblastoma | Increase in expression of miRNA within the miR-379/miR-656 cluster post-dCas9-VP64 gene induction. |
| miR-23b and miR-27b [186] | Annotated Stem-loop region | Cas9/Lentiviral Transduction | Human Breast Cancer (MCF7) | Significant reduction of miR-23b and miR-27b transcripts was observed |
| miR-423 [187] | miR-423 locus | Cas9/Lipofection | Human Cervical Cancer (HeLa) | Significant knockdown of miR-423 transcripts was observed |
| miR-17-92 [188] | miR-17-92 5p loop | CRISPR/Cas9 nickases | Anaplastic Thyroid Cancer | Knockdown of clusters was observed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azlan, A.; Rajasegaran, Y.; Kang Zi, K.; Rosli, A.A.; Yik, M.Y.; Yusoff, N.M.; Heidenreich, O.; Moses, E.J. Elucidating miRNA Function in Cancer Biology via the Molecular Genetics’ Toolbox. Biomedicines 2022, 10, 915. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10040915
Azlan A, Rajasegaran Y, Kang Zi K, Rosli AA, Yik MY, Yusoff NM, Heidenreich O, Moses EJ. Elucidating miRNA Function in Cancer Biology via the Molecular Genetics’ Toolbox. Biomedicines. 2022; 10(4):915. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10040915
Chicago/Turabian StyleAzlan, Adam, Yaashini Rajasegaran, Khor Kang Zi, Aliaa Arina Rosli, Mot Yee Yik, Narazah Mohd Yusoff, Olaf Heidenreich, and Emmanuel Jairaj Moses. 2022. "Elucidating miRNA Function in Cancer Biology via the Molecular Genetics’ Toolbox" Biomedicines 10, no. 4: 915. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines10040915