
Improving the Treatment Efficiency and Lowering the Operating Costs of Electrochemical Advanced Oxidation Processes

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Artificial Wastewater
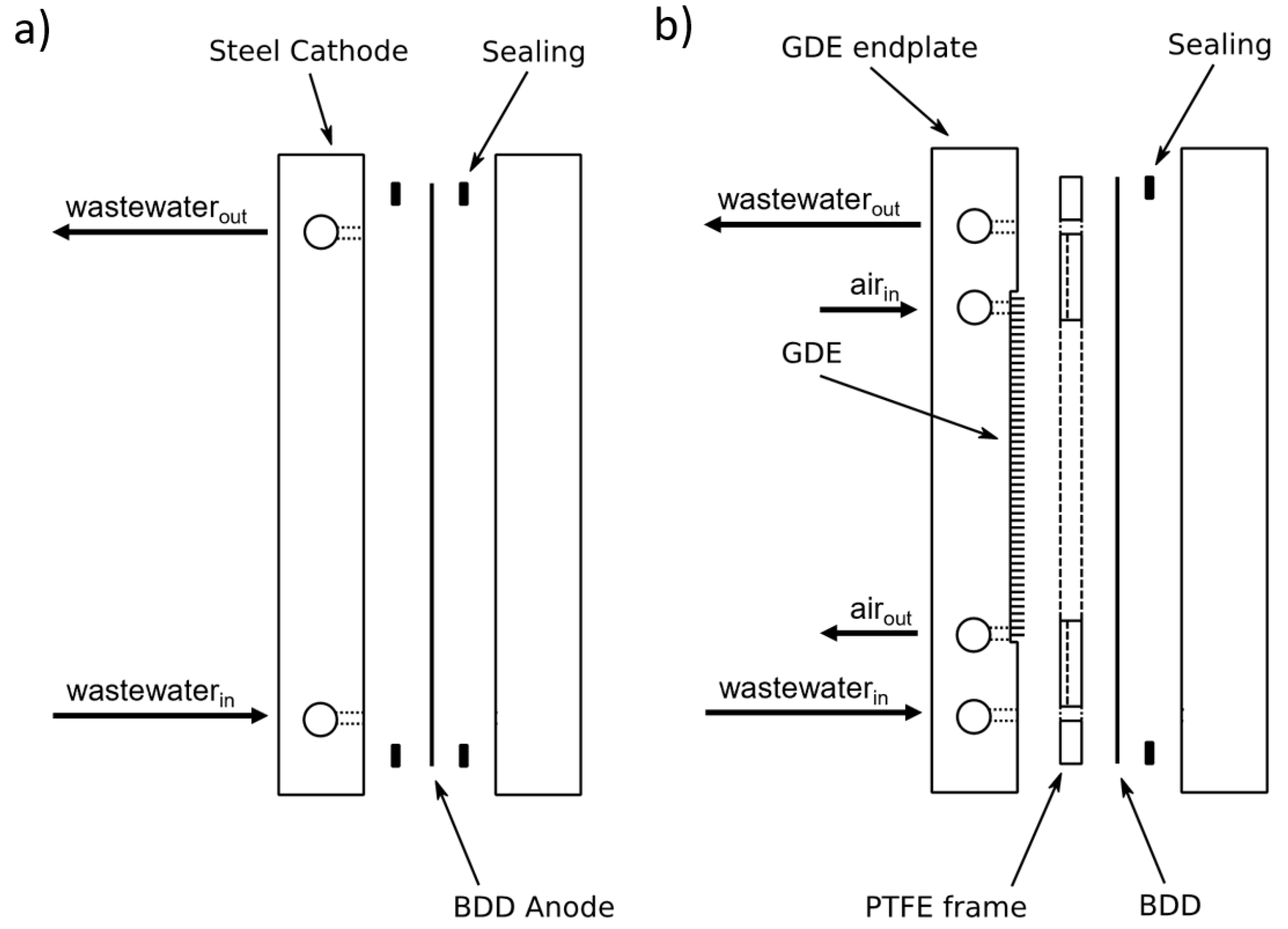
2.2. Electrolysis Setups
2.3. Ozone and Peroxone Process
2.4. Analytical Methods and Performance Data
3. Results
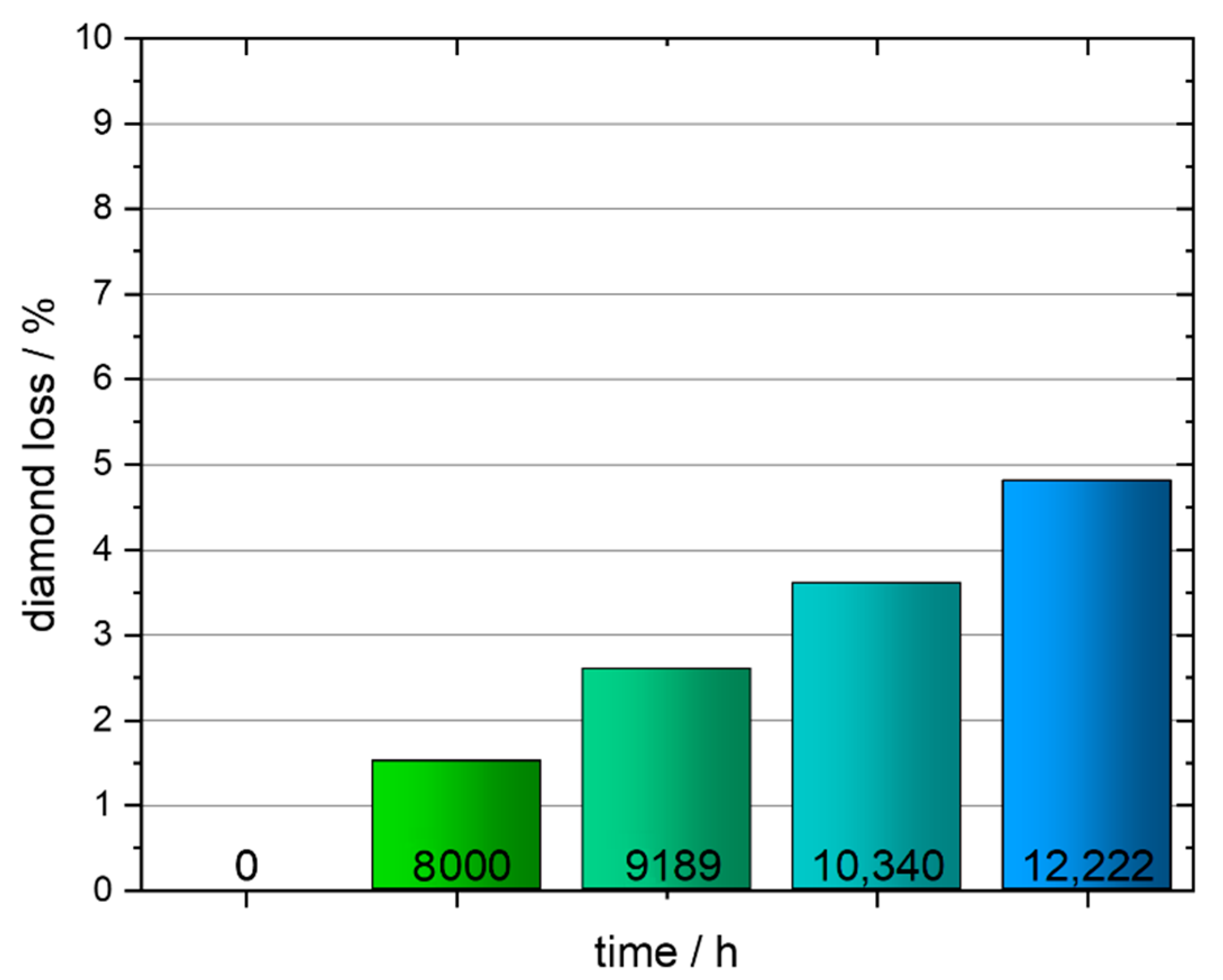
3.1. BDD Coating Thickness
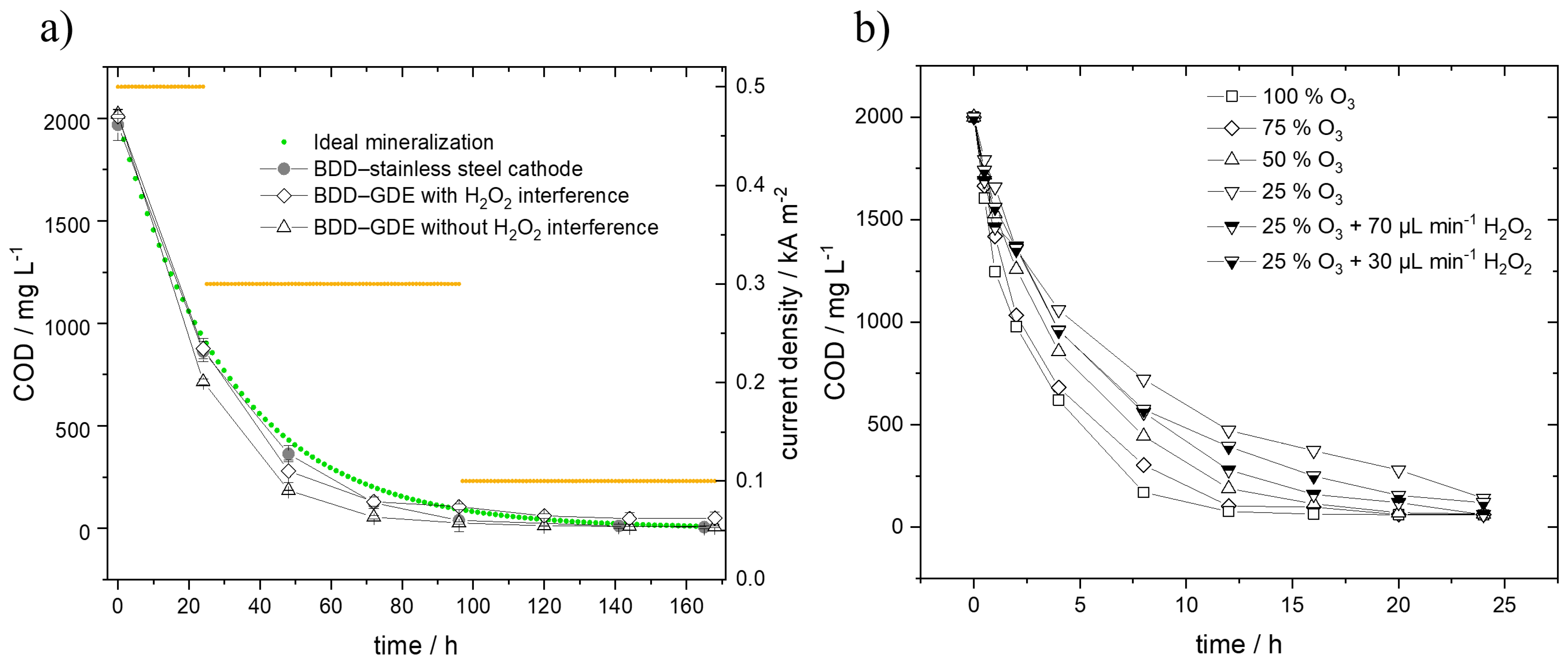
3.2. Treatment Performance of the Electrolysis and the Ozone and Peroxone Processes
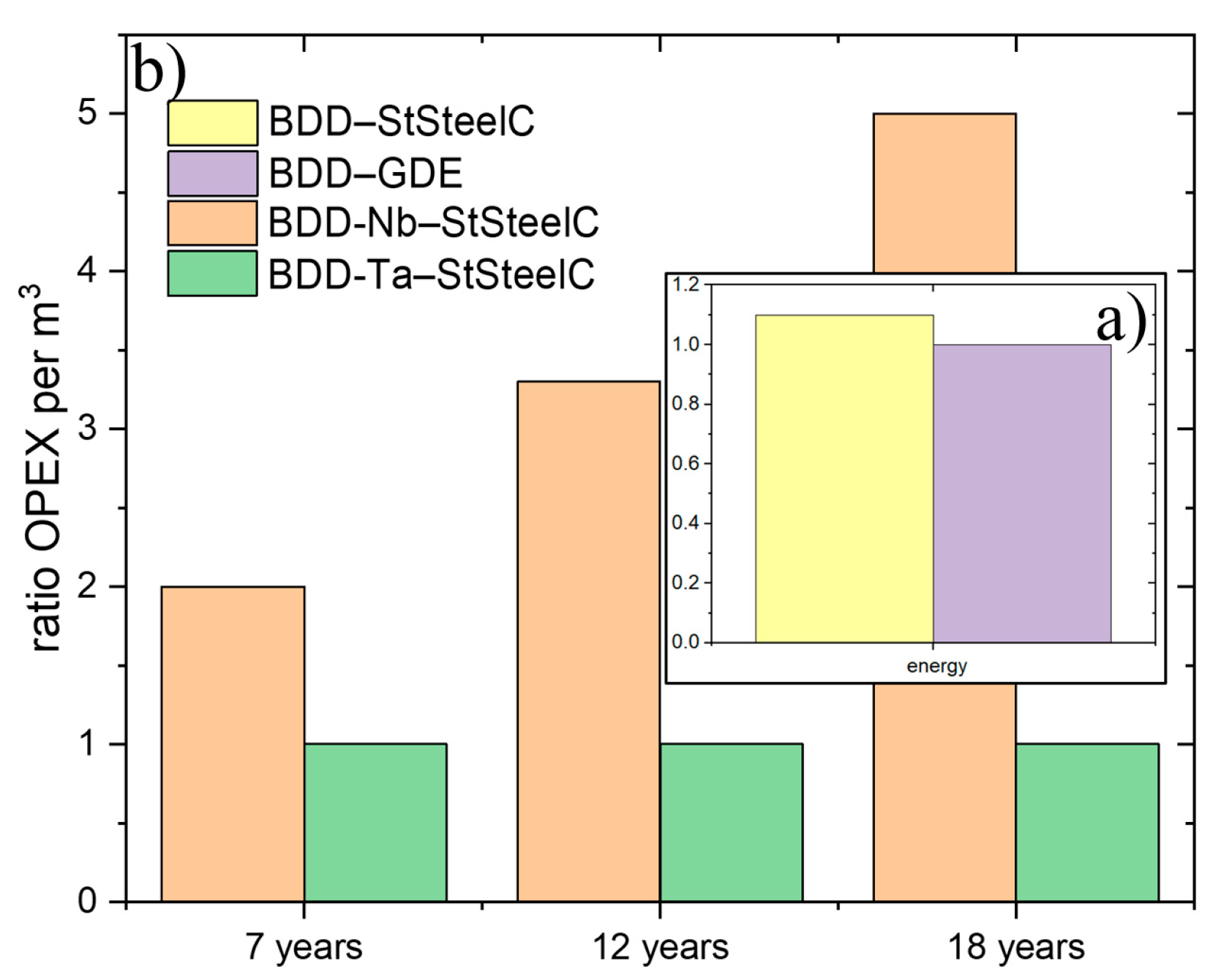
3.3. Operational Expense of Ta-BDD-Based EAOP®
- 2.5 m3 h−1 WW from pharmaceutical production;
- COD degradation from 2 g L−1 to 0.02 g L−1;
- 7, 12, or 18 year lifetime for tantalum-based DIACHEM® electrodes, based on the BDD coating thickness investigation carried out in this study;
- 2 year lifetime for niobium-based DIACHEM® electrodes as the reference material, which is the previous standard material for BDD electrodes, based on our industrial experience, which is consistent with Comninellis et al. [49];
- 0.1 € kWh−1 energy costs;
- Specific charge of degradation with cell BDD–stainless steel process is equal to specific charge of degradation with cell BDD–GDE process;
- Adapted current density from 0.5 kA m−2 to 0.1 kA m−2 as performed during the investigations.
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martínez-Huitle, C.A.; Panizza, M. Electrochemical oxidation of organic pollutants for wastewater treatment. Curr. Opin. Electrochem. 2018, 11, 62–71. [Google Scholar] [CrossRef]
- Särkkä, H.; Bhatnagar, A.; Sillanpää, M. Recent developments of electro-oxidation in water treatment—A review. J. Electroanal. Chem. 2015, 754, 46–56. [Google Scholar] [CrossRef]
- Ganiyu, S.O.; Martínez-Huitle, C.A.; Rodrigo, M.A. Renewable energies driven electrochemical wastewater/soil decontamination technologies: A critical review of fundamental concepts and applications. Appl. Catal. Environ. 2020, 270, 118857. [Google Scholar] [CrossRef]
- Ganiyu, S.O.; Martínez-Huitle, C.A.; Oturan, M.A. Electrochemical advanced oxidation processes for wastewater treatment: Advances in formation and detection of reactive species and mechanisms. Curr. Opin. Electrochem. 2020, 27, 100678. [Google Scholar] [CrossRef]
- Jiang, H.; Dang, C.; Liu, W.; Wang, T. Radical attack and mineralization mechanisms on electrochemical oxidation of p-substituted phenols at boron-doped diamond anodes. Chemosphere 2020, 248, 126033. [Google Scholar] [CrossRef] [PubMed]
- Muddemann, T.; Haupt, D.; Sievers, M.; Kunz, U. Elektrochemische Reaktoren für die Wasserbehandlung. Chem. Ing. Tech. 2019, 91, 769–785. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.G.; Stöckl, M.; Becker, D.; Steinkamp, A.-D.; Abt, C.; Jungfer, C.; Weidlich, C.; Track, T.; Mangold, K.-M. Current to clean water—Electrochemical solutions for groundwater, water, and wastewater treatment. Chem. Ing. Tech. 2018, 90, 1832–1854. [Google Scholar] [CrossRef]
- Ganiyu, S.O.; Martínez-Huitle, C.A. The use of renewable energies driving electrochemical technologies for environmental applications. Curr. Opin. Electrochem. 2020, 22, 211–220. [Google Scholar] [CrossRef]
- Radjenovic, J.; Sedlak, D.L. Challenges and opportunities for electrochemical processes as next-generation technologies for the treatment of contaminated water. Environ. Sci. Technol. 2015, 49, 11292–11302. [Google Scholar] [CrossRef] [PubMed]
- Barndõk, H.; Hermosilla, D.; Negro, C.; Blanco, A.M. Comparison and predesign cost assessment of different advanced oxidation processes for the treatment of 1,4-dioxane-containing wastewater from the chemical industry. ACS Sustain. Chem. Eng. 2018, 6, 5888–5894. [Google Scholar] [CrossRef]
- He, Y.; Lin, H.; Guo, Z.; Zhang, W.; Li, H.; Huang, W. Recent developments and advances in boron-doped diamond electrodes for electrochemical oxidation of organic pollutants. Sep. Purif. Technol. 2018, 212, 802–821. [Google Scholar] [CrossRef]
- Muddemann, T.; Bulan, A.; Sievers, M.; Kunz, U. Avoidance of chlorine formation during electrolysis at boron-doped diamond anodes in highly sodium chloride containing and organic-polluted wastewater. J. Electrochem. Soc. 2018, 165, J3281–J3287. [Google Scholar] [CrossRef]
- Muddemann, T.; Haupt, D.; Sievers, M.; Kunz, U. Electrochemical reactors for wastewater treatment. Chem. Biol. Eng. Rev. 2019, 6, 142–156. [Google Scholar] [CrossRef]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef]
- Giomo, M.; Buso, A.; Fier, P.; Sandona, G.; Boye, B.; Farnia, G. A small-scale pilot plant using an oxygen-reducing gas-diffusion electrode for hydrogen peroxide electrosynthesis. Electrochim. Acta 2008, 54, 808–815. [Google Scholar] [CrossRef]
- Elvers, B. Ullmann’s Encyclopedia of Industrial Chemistry; Chemie: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Busca, G.; Berardinelli, S.; Resini, C.; Arrighi, L. Technologies for the removal of phenol from fluid streams: A short review of recent developments. J. Hazard. Mater. 2008, 160, 265–288. [Google Scholar] [CrossRef]
- Moussallem, I.; Jörissen, J.; Kunz, U.; Pinnow, S.; Turek, T. Chlor-alkali electrolysis with oxygen depolarized cathodes: History, present status and future prospects. J. Appl. Electrochem. 2008, 38, 1177–1194. [Google Scholar] [CrossRef]
- Cornejo, O.M.; Murrieta, M.F.; Castañeda, L.F.; Nava, J.L. Characterization of the reaction environment in flow reactors fitted with BDD electrodes for use in electrochemical advanced oxidation processes: A critical review. Electrochim. Acta 2019, 331, 135373. [Google Scholar] [CrossRef]
- Dominguez-Ramos, A.; Aldaco, R.; Irabien, A. Photovoltaic solar electrochemical oxidation (PSEO) for treatment of lignosulfonate wastewater. J. Chem. Technol. Biotechnol. 2010, 85, 821–830. [Google Scholar] [CrossRef]
- Luo, H.; Li, C.; Wu, C.; Zheng, W.; Dong, X. Electrochemical degradation of phenol by in situ electro-generated and electro-activated hydrogen peroxide using an improved gas diffusion cathode. Electrochim. Acta 2015, 186, 486–493. [Google Scholar] [CrossRef]
- Agladze, G.R.; Tsurtsumia, G.S.; Jung, B.-I.; Kim, J.-S.; Gorelishvili, G. The “In-cell” and “Ex-cell” Fenton treatment of phenol, 4-chlorophenol and aniline. J. Appl. Electrochem. 2007, 37, 385–393. [Google Scholar] [CrossRef]
- Huang, B.-C.; Jiang, J.; Wang, W.-K.; Li, W.-W.; Zhang, F.; Jiang, H.; Yu, H.-Q. Electrochemically catalytic degradation of phenol with hydrogen peroxide in situ generated and activated by a municipal sludge-derived catalyst. ACS Sustain. Chem. Eng. 2018, 6, 5540–5546. [Google Scholar] [CrossRef]
- Abbas, Z.I.; Abbas, A. Oxidative degradation of phenolic wastewater by electro-fenton process using MnO2-graphite electrode. J. Environ. Chem. Eng. 2019, 7, 103108. [Google Scholar] [CrossRef]
- Ma, H.; Zhang, X.; Ma, Q.; Wang, B. Electrochemical catalytic treatment of phenol wastewater. J. Hazard. Mater. 2009, 165, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Xu, F.; Gao, J.; Yuen, M.; Sun, S.; Xu, J.; Jia, K.; Zuo, D. Nanostructured boron-doped diamond electrode for degradation of the simulation wastewater of phenol. Diam. Relat. Mater. 2020, 109, 108098. [Google Scholar] [CrossRef]
- Gui, L.; Chen, Z.; Chen, B.; Song, Y.; Yu, Q.; Zhu, W.; Hu, Q.; Liu, Y.; Zheng, Z.; Ze, L.; et al. Preparation and characterization of ZnO/PEG-Co(II)-PbO2 nanocomposite electrode and an investigation of the electrocatalytic degradation of phenol. J. Hazard. Mater. 2020, 399, 123018. [Google Scholar] [CrossRef]
- Li, X.-Y.; Cui, Y.-H.; Feng, Y.; Xie, Z.-M.; Gu, J.-D. Reaction pathways and mechanisms of the electrochemical degradation of phenol on different electrodes. Water Res. 2005, 39, 1972–1981. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Zhou, M. Partial degradation of phenol by advanced electrochemical oxidation process. Environ. Sci. Technol. 2001, 35, 2698–2703. [Google Scholar] [CrossRef] [PubMed]
- Haupt, D.; Muddemann, T.; Kunz, U.; Sievers, M. Evaluation of a new electrochemical concept for vacuum toilet wastewater treatment—Comparison with ozonation and peroxone processes. Electrochem. Commun. 2019, 101, 115–119. [Google Scholar] [CrossRef]
- Suzuki, H.; Araki, S.; Yamamoto, H. Evaluation of advanced oxidation processes (AOP) using O3, UV, and TiO2 for the degradation of phenol in water. J. Water Process. Eng. 2015, 7, 54–60. [Google Scholar] [CrossRef]
- Panizza, M.; Sineé, G.; Duo, I.; Ouattara, L.; Comninellis, C. Electrochemical polishing of boron-doped diamond in organic media. Electrochem. Solid State Lett. 2003, 6, D17. [Google Scholar] [CrossRef]
- de Araújo, D.M.; Sáez, C.; Cañizares, P.; Rodrigo, M.A.; Martínez-Huitle, C.A. Improving the catalytic effect of peroxodisulfate and peroxodiphosphate electrochemically generated at diamond electrode by activation with light irradiation. Chemosphere 2018, 207, 774–780. [Google Scholar] [CrossRef] [Green Version]
- Serrano, K.G. A critical review on the electrochemical production and use of peroxo-compounds. Curr. Opin. Electrochem. 2020, 27, 100679. [Google Scholar] [CrossRef]
- Liu, H.; Bruton, T.A.; Doyle, F.M.; Sedlak, D.L. In situ chemical oxidation of contaminated groundwater by persulfate: Decomposition by Fe(III) and Mn(IV) containing oxides and aquifer materials. Environ. Sci. Technol. 2014, 48, 10330–10336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assumpção, M.; De Souza, R.; Rascio, D.; Silva, J.C.; Calegaro, M.; Gaubeur, I.; Paixao, T.; Hammer, P.; Lanza, M.; Santos, M. A comparative study of the electrogeneration of hydrogen peroxide using Vulcan and Printex carbon supports. Carbon 2011, 49, 2842–2851. [Google Scholar] [CrossRef]
- Barros, W.; Wei, Q.; Zhang, G.; Sun, S.; Lanza, M.; Tavares, A.C. Oxygen reduction to hydrogen peroxide on Fe3O4 nanoparticles supported on Printex carbon and Graphene. Electrochim. Acta 2015, 162, 263–270. [Google Scholar] [CrossRef]
- Fryda, M.; Matthée, T.; Mulcahy, S.; Hampel, A.; Schäfer, L.; Tröster, I. Fabrication and application of Diachem electrodes. Diam. Relat. Mater. 2003, 12, 1950–1956. [Google Scholar] [CrossRef]
- Neuber, R.; Grassl, T.; Matthée, T. Elektrochemische Zelle. EP3816322, 29 October 2020. [Google Scholar]
- Lee, E.; Lee, H.; Kim, Y.K.; Sohn, K.; Lee, K. Hydrogen peroxide interference in chemical oxygen demand during ozone based advanced oxidation of anaerobically digested livestock wastewater. Int. J. Environ. Sci. Technol. 2011, 8, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Issa, M.; Muddemann, T.; Haupt, D.; Kunz, U.; Sievers, M. Simple catalytical approach for removal of analytical interferences caused by hydrogen peroxide in a standard chemical oxygen demand test. J. Environ. Eng. 2021. [Google Scholar] [CrossRef]
- Muddemann, T.; Haupt, D.R.; Sievers, M.; Kunz, U. Improved operating parameters for hydrogen peroxide-generating gas diffusion electrodes. Chem. Ing. Tech. 2020, 92, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Panizza, M.; Brillas, E.; Comninellis, C. Application of boron-doped diamond electrodes for wastewater treatment. J. Environ. Eng. Manag. 2008, 18, 139–153. [Google Scholar]
- Moradi, M.; Vasseghian, Y.; Khataee, A.; Kobya, M.; Arabzade, H.; Dragoi, E.-N. Service life and stability of electrodes applied in electrochemical advanced oxidation processes: A comprehensive review. J. Ind. Eng. Chem. 2020, 87, 18–39. [Google Scholar] [CrossRef]
- Ginovska, B.; Dupuis, M.; Camaioni, D.M. The H2O2+OH→HO2+H2O reaction in aqueous solution from a charge-dependent continuum model of solvation. J. Chem. Phys. 2008, 129, 014506. [Google Scholar] [CrossRef]
- Verordnung über Anforderungen an das Einleiten von Abwasser in Gewässer (Abwasserverordnung-AbwV). Available online: https://www.gesetze-im-internet.de/abwv/AbwV.pdf (accessed on 29 July 2021).
- Sievers, M. Advanced Oxidation Processes. In Treatise of Water Science; Wilderer, P., Ed.; Elsevier: Oxford, UK, 2011. [Google Scholar]
- Haynes, W.M. CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Physical Data, 97th ed.; CRC Press: London, UK, 2017. [Google Scholar]
- Kraft, A. Doped diamond: A compact review on a new, versatile electrode material. Int. J. Electrochem. Sci. 2007, 2, 355–385. [Google Scholar]
- Duan, X.; Ma, F.; Yuan, Z.; Chang, L.; Jin, X. Electrochemical degradation of phenol in aqueous solution using PbO2 anode. J. Taiwan Inst. Chem. Eng. 2012, 44, 95–102. [Google Scholar] [CrossRef]
- Jin, P.; Chang, R.; Liu, D.; Zhao, K.; Zhang, L.; Ouyang, Y. Phenol degradation in an electrochemical system with TiO2/activated carbon fiber as electrode. J. Environ. Chem. Eng. 2014, 2, 1040–1047. [Google Scholar] [CrossRef]
- García-García, A.; Martínez-Miranda, V.; Martínez-Cienfuegos, I.G.; Almazán-Sánchez, P.T.; Castañeda-Juárez, M.; Linares-Hernández, I. Industrial wastewater treatment by electrocoagulation-electrooxidation processes powered by solar cells. Fuel 2015, 149, 46–54. [Google Scholar] [CrossRef]
- Henrique, J.; De Andrade, D.C.; Neto, E.L.B.; Da Silva, D.R.; Dos Santos, E.V. Solar-powered BDD-electrolysis remediation of soil washing fluid spiked with diesel. J. Chem. Technol. Biotechnol. 2019, 94, 2999–3006. [Google Scholar] [CrossRef]
- Souza, F.; Lanza, M.; Llanos, J.; Sáez, C.; Rodrigo, M.A.; Cañizares, P. A wind-powered BDD electrochemical oxidation process for the removal of herbicides. J. Environ. Manag. 2015, 158, 36–39. [Google Scholar] [CrossRef] [PubMed]







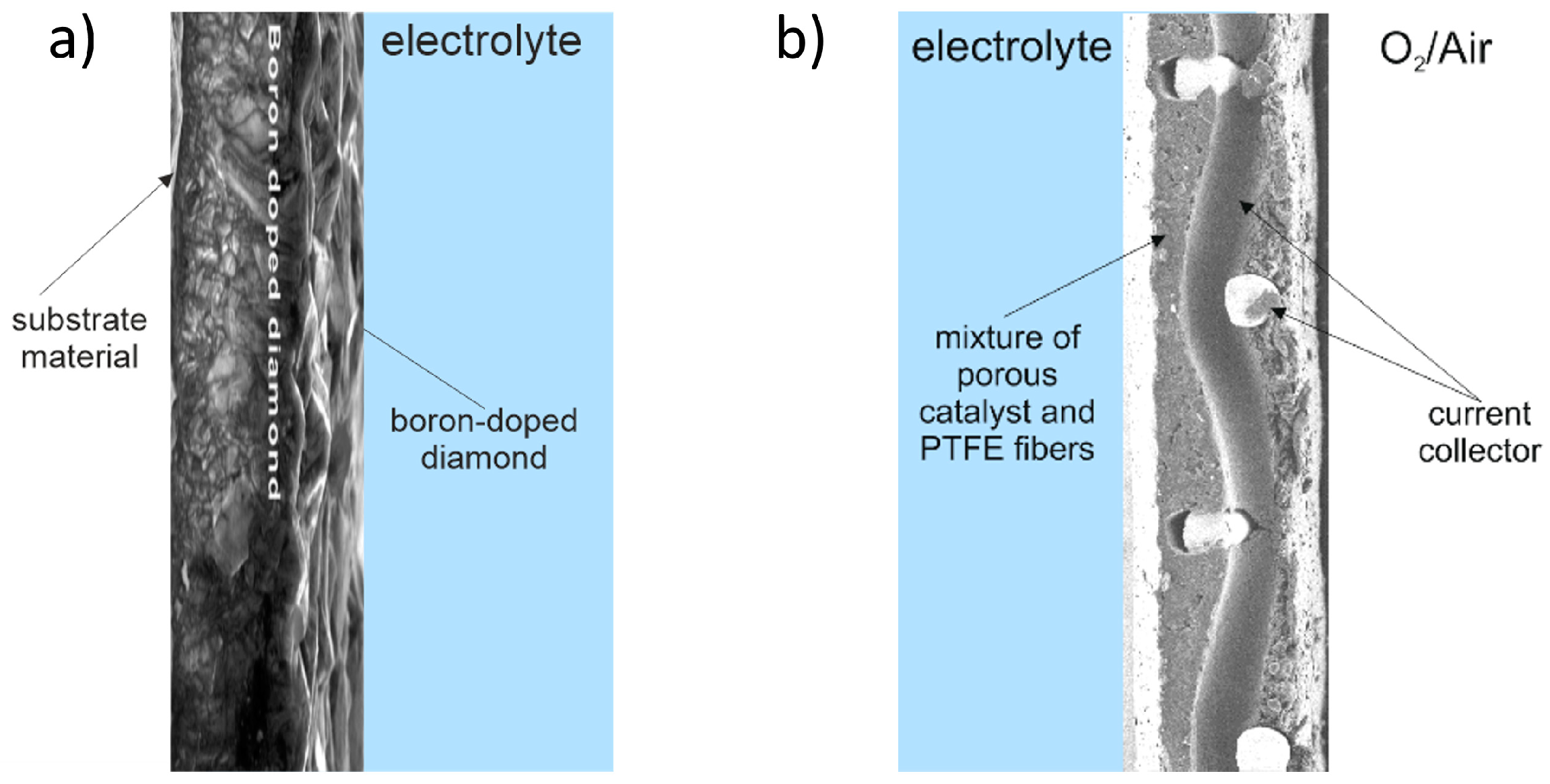{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode Configuration | Energy Demand Over 6 Days | Energy Demand Reaching Concentration of Discharge | ||
|---|---|---|---|---|
/kWh m−3 | /kWh kgCOD−1 | /kWh m−3 | /kWh kgCOD−1 | |
| BDD–stainless steel cathode | 87.57 ± 3.91 | 43.79 ± 1.95 | 78.89 ± 3.84 | 39.45 ± 1.92 |
| BDD–GDE | 80.46 ± 0.73 | 40.23 ± 0.37 | 58.84 ± 1.18 | 29.42 ± 0.59 |
| Power Setting (O3 Generator) and Optional H2O2 Dosing (Peroxone) /% | /W | /kWh kgO3−1 | /kWh kgH2O2−1 | /mgO3 mgCOD−1 | /kWh kgCOD−1 |
|---|---|---|---|---|---|
| Idle | 17.6 | 0 | - | - | - |
| 25% | 30.4 | 16.5 | - | 11.9 | 196.3 |
| 50% | 47 | 20.6 | - | 14.1 | 291.5 |
| 75% | 61.5 | 22.9 | - | 16.6 | 380.8 |
| 100% | 76 | 24.1 | - | 19.5 | 470.0 |
| 25% + 0.03 mL min−1 | - | 16.5 | 15 | 11.74 * | 349.0 |
| 25% + 0.07 mL min−1 | - | 16.5 | 15 | 11.39 * | 539.0 |
| Main Electrode | Special Feature | cPhenol [mg L−1] | Na2SO4 [g L−1] | Mineralization Rate [%] | CE [%] | Specific Energy per Mass COD or TOC [kWh kgCOD/TOC−1] | Ref. |
|---|---|---|---|---|---|---|---|
| C | Generated and activated H2O2 | 50 | 28.4 | 85 (TOC) | - | - | [21] |
| C | In-cell and ex-cell Fenton | 282 | 1 | 90 (COD) | - | 56.3 | [22] |
| C | Generated and activated H2O2 | 40 | 7.1 | 87 (TOC) | - | - | [23] |
| C | Electrochemical catalytic treatment | 520 | 4 | 99 (COD) | - | - | [25] |
| A | Nanostructured BDD | 9153 | 49 g L−1 H2SO4 | 50 (TOC) | [26] | ||
| A | Modified PbO2-based anode | 100 | 14.2 | 91.1 (phenol) | - | - | [27] |
| A | PbO2 anode | 50 | 7.1 | 94 (TOC) | 1.8 | 3759 | [50] |
| A | TiO2 with activated carbon fiber | 100 | 10.7 | 83.3 (phenol) | - | - | [51] |
| A | BDD anode | 850 | 9 | >99 (COD) | ≈100 | 43.79 | This study |
| A+C | BDD anode + H2O2 GDE | 850 | 9 | >99 (COD) | ≈100 | 40.23 | This study |
| Main Electrode | Regenerative Energy | Type of ww | Mineralization [%] | [kWh kgCOD−1] | [kWh m−3] | Ref. |
|---|---|---|---|---|---|---|
| BDD | PV | Industrial | 99 (TOC) | - | 896 | [52] |
| BDD | PV | lignosulfonate | 90 (TOC) | - | 100 | [20] |
| BDD | PV | Soil washing fluid spiked with diesel | 57–76 (TOC) | 2300 | - | [53] |
| BDD | WT | herbicides | >99 (TOC) | - | 18.75 kAh m−3 | [54] |
| BDD | - | Phenolic | >99 (COD) | 43.79 | 87.57 | This study |
| BDD + H2O2-GDE | - | Phenolic | >99 (COD) | 40.23 | 80.46 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muddemann, T.; Neuber, R.; Haupt, D.; Graßl, T.; Issa, M.; Bienen, F.; Enstrup, M.; Möller, J.; Matthée, T.; Sievers, M.; et al. Improving the Treatment Efficiency and Lowering the Operating Costs of Electrochemical Advanced Oxidation Processes. Processes 2021, 9, 1482. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9091482
Muddemann T, Neuber R, Haupt D, Graßl T, Issa M, Bienen F, Enstrup M, Möller J, Matthée T, Sievers M, et al. Improving the Treatment Efficiency and Lowering the Operating Costs of Electrochemical Advanced Oxidation Processes. Processes. 2021; 9(9):1482. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9091482
Chicago/Turabian StyleMuddemann, Thorben, Rieke Neuber, Dennis Haupt, Tobias Graßl, Mohammad Issa, Fabian Bienen, Marius Enstrup, Jonatan Möller, Thorsten Matthée, Michael Sievers, and et al. 2021. "Improving the Treatment Efficiency and Lowering the Operating Costs of Electrochemical Advanced Oxidation Processes" Processes 9, no. 9: 1482. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9091482