1. The History of Molecular Magnetism in Brief—The Era of Single-Molecule Magnets (SMMs) and Single-Ion Magnets (SIMs)
Molecular magnetism [
1] is currently a “hot” interdisciplinary research field which started almost 35 years ago. It was a rather natural extension of the field of Magnetochemistry which was developed in the 1955–1985 period. In the latter field, inorganic chemists were using experimental values of magnetic moment and their variation with temperature to draw chemical and structural conclusions for molecular complexes. Well known examples [
2,
3] were: (i) The distinction between several stereochemistries (tetrahedral, square planar, octahedral) in complexes of some 3d-metal ions, e.g., Ni(II) and Co(II); (ii) the recognition of high-spin and low-spin configurations in octahedral complexes of 3d
4–3d
7 metal ions; and (iii) the elegant determination of the singlet-triplet gap in copper(II) acetate hydrate by Bleaney and Bowers, before the X-ray solution of its dinuclear [Cu
2(O
2CMe)
4(H
2O)
2] structure.
Before proceeding to a brief history of molecular magnetism, we can mention two advantages that molecular systems offer over conventional atom- or ion-based magnetic materials [
4]: (a) The structures that can be formed are more complicated and more diverse that the structural types we normally meet in conventional inorganic materials. Molecular crystals can provide scientists with ensembles of identical structures and iso-orientated magnetic objects which permit the in-depth study of their physical behavior. Such molecular materials offer model systems to test existing theories on many-body problems and discover, e.g., new quantum, behavior; and (b) Molecular systems are ideal for incorporation of other useful functionalities, e.g., optical properties, conductivity, etc. This may be a synergistic property not coupled with magnetism, or it may be a coupling of different physical properties, e.g., light-induced magnetic ordering in spin-crossover (SCO) Prussian blue phases. This type of materials may find novel, highly specific applications.
In the field of molecular magnetism, chemists, chemical engineers, physicists and material scientists, both experimentalists and theoreticians, closely collaborate trying to design, synthesize, fully characterize and model the magnetic properties of molecule-based materials. In the initial period of its development, the focus of research was on simple model systems (homo- and heterometallic dinuclear complexes and coordination clusters); the goal was to test theories in solids about exchange interactions and electron delocalization at the molecular scale [
5]. The field later expanded towards the study of 1D systems [
1,
6,
7], e.g., homometallic chains, heterometallic chains and homometallic chains in which the metal centers are bridged by an organic radical. In the late 1980s and during the 1990s, chemists prepared a great variety of 3D complexes exhibiting spontaneous magnetization below a critical temperature (
Tc) [
8,
9,
10]. The Miller and Epstein groups broke the critical temperature barrier record (
Tc > 350 K) with the ferrimagnet {[V
III(TCNE)
2]∙
xCH
2Cl
2}
n [
11], where TCNE
− is the radical anion of tetracyanoethylene, while other groups came close by using Prussian blue derivatives [
9,
10].
Simultaneously with the above developments, the molecular magnetism community paid more attention on octahedral 3d
4–3d
7 systems (mainly on 3d
6 iron(II) complexes) with a SCO behavior [
12,
13]. This phenomenon, discovered by Cambi in 1931, continues to attract the intense interest of scientists even after 90 years [
14,
15]. The goals are to construct systems which undergo spin transition near 300 K and to study the possibility of tuning this molecular bistability through application of external stimuli (temperature, pressure, light). The SCO phenomenon has already led to applications, e.g., sensors [
16], actuators [
17] and thermal displays [
18].
Thus, the initial growth of molecular magnetism was mainly based on the deep understanding of the factors that could be used to design and synthesize novel crystalline molecule-based materials exhibiting useful magnetic properties, e.g., ferromagnetism and ferrimagnetism, similar to those observed in inorganic atom-/ion-based materials. More importantly, those molecular materials had special features such as low density, insulating nature and optical transparency; these distinctive physical properties provide material scientists with a number of fabrication advantages, as the materials are most often prepared using solution methods.
After the period of initial growth of molecular magnetism, its community turned the attention to three large classes of molecular compounds. The first class comprises the Single-Molecule Magnets (SMMs) and the Single-Ion Magnets (SIMs), the second the Single-Chain Magnets (SCMs) and the third involves the multifunctional molecular magnetic materials [
19,
20]. The focus of this review is on transition-metal SMMs and SIMs.
In the presence of axial magnetic anisotropy (
D), the
MS levels of a transition-metal complex with total spin
S will split under zero magnetic field according to the Hamiltonian
Ĥ = DŜz2. If the value of
D is negative, the two ±
MS levels of maximal projection along the
z axis form a bistable ground state because they are degenerate. If we reverse the magnetic moment by converting −
MS to +
MS, this requires traversal of a spin-inversion barrier. This barrier is
U =
S2|
D| for integer
S or
U = (
S2 − 1/4)|
D| for non-integer
S, and the system passes through the
MS = 0 or the
MS = 1/2 levels, respectively, at the height of the energy barrier. The existence of such a barrier can lead to the slow relaxation of the magnetic moment at very low temperatures upon removal of the external dc field [
21,
22]. The presence of this barrier is often proven by the appearance of magnetic hysteresis of molecular origin as first observed for the iconic [Mn
12O
12(O
2CMe)
16(H
2O)
4] (
1,
Figure 1) SMM [
21,
22,
23]. Clusters containing polynuclear molecules that exhibit such behavior have been named Single-Molecule Magnets. The magnetic behavior of each of these clusters can be described as a giant anisotropic spin as a result of the exchange coupling between the spins of neighboring metal ions. Because of the magnetic bistability, these polynuclear molecules were proposed for use in magnetic memory devices since they can remain magnetized in one of the two spin states, thus giving rise to a “bit” of memory. The aim during the first decade of SMM research (1993–2003) was to prepare SMMs with memory effects at higher temperatures [
19]. Although synthetic inorganic chemists made many efforts to achieve this goal, the progress was little and the energy barriers that stabilize the magnetic bits against thermal fluctuations remained small. Another tremendously important consequence of the discovery of SMMs was the observation of quantum effects in mesoscopic magnets. At that time, physicists were looking for small magnetic particles, all identical to each other, to investigate if quantum effects could be observed in ensembles of such identical particles; however, the preparation of these collections proved difficult. Chemists solved the problem using a molecular approach to prepare identical cluster molecules in crystalline SMMs. A few years after the characterization of
1, scientists revealed that its crystals exhibit quantum tunneling of magnetization (QTM) [
24,
25]; this phenomenon is considered one of the milestones in the study of spin during the 20th century [
19]. Synthetic efforts were followed by advanced theoretical studies, and the latter provided strong evidence that the magnitude of
D decreases as
S increases; this implied that the construction of efficient SMMs with a large
U cannot be achieved by only maximizing
S and that control of
D is equally important [
21,
22].
In the last 15 years or so, another exciting subclass of SMMs was discovered, the so-called Single-Ion Magnets (SIMs). These represent the smallest molecular nanomagnets we can imagine [
21,
26,
27,
28,
29,
30,
31,
32,
33,
34]. In the literature, SIMs are often referred to as mononuclear SMMs. However, neither SIMs nor mononuclear SMMs are perfect descriptors [
26]. Monometallic SMMs is probably better, but herein we have chosen to use the SIM acronym rather, e.g., MSMM, which is probably more awkward. SIMs are mononuclear complexes containing a single magnetic d- or f-metal ion. The motivation behind the SIM research evolution was the belief of scientists that incorporating many paramagnetic centers into a cluster molecule may be a disadvantage in terms of generating complexes with large cluster anisotropies (
D or better
Dcluster). Many metal ions can, in principle, lead to a large
S, but as the nuclearity of the molecule increases it is becoming very difficult (practically impossible) to control the mutual alignment of the anisotropy axes of the individual metal ions; this gives small
Dcluster values [
27]. These considerations, combined with the observation of slow magnetization relaxation in complexes (Bu
n4N)[Ln(pc)
2] (
2), where Ln = Tb, Dy and pc = the dianion of phthalocyanine [
35], by Ishikawa and co-workers, turned the attention of researchers to single-ion systems [
19,
21]. If we compare 4f-(Ln) and d-metal ions for use in SIMs, the former appear better because they possess: (i) larger magnetic moments, (ii) higher spin-orbit coupling constants, and (iii) weak coupling of the f orbitals to the ligand field which cannot quench first-orbital contributions to the magnetic moment. As a consequence, magnetic hysteresis has been observed at temperatures as high as 80 K for mononuclear organometallic Dy(III) complexes [
36]. However, d-metal ion SIMs are important and cannot be considered simply as academic research curiosities. As scientists understand deeper the physics of d single ions in a ligand field, they can begin to design strategies to couple the anisotropy of individual transition metal ions together in order to create cluster SMMs in a rational way; such clusters could be ideal molecular analogues for magnetic nanoparticles. Additionally, the utilization of well-studied and understood SIM building blocks in the modular synthesis of 1D SCMs with high spin and uniaxial anisotropies (a concept which is active in the 4f-metal chemistry), and Metal–Organic Frameworks (MOFs) with SIM units as nodes (e.g., for the design of porous magnetic materials) is a currently “hot” research area, see information provided in reference [
27].
Despite high energy barriers for magnetization reversal, often SMMs and SIMs suffer from fast relaxation processes, not just via QTM, but also via interactions between the spin states and lattice phonons [
27,
30]. The complex behavior of relaxation dynamics is a specialized topic beyond the scope of this review. However, it is helpful to consider the SMM and SIM systems as being composed of two parts, the spin part and the lattice part. The interaction between lattice vibrations (phonons) and spin provide the system with additional relaxation processes, which “shortcut” the desired thermal pathway. The spin-lattice relaxation mechanisms are of three types (direct, Orbach and Raman processes). Since the tunneling pathways are very sensitive to changes of molecular symmetry, synthetic chemists try to control the molecular symmetry of SMMs/SIMs through careful design of the ligands used. Additional tools for minimizing QTM through the ground state are magnetic dilution and utilization of a Kramers metal ion; for the latter, breaking of the
MS degeneracy and therefore QTM is formally forbidden in strictly zero magnetic field. However, the fast quantum tunneling offers an advantage because these molecules are candidates to realize quantum bits (qubits), the basic units of quantum computers [
37].
A second class of molecular compounds which has captured the intense interest of the molecular magnetism community consists of slow-relaxing 1D magnets, or SCMs [
20,
21,
38,
39]. These consist of chains, isolated from each other, presenting a slow relaxation of the magnetization; they cannot present a long-range magnetic order. However, they exhibit a short-range order caused by the occurrence of domains where N spins are oriented in the same direction, interrupted by a reversed spin or by chain defects. A finite magnetization can thus be frozen in the absence of an applied magnetic field at low temperatures. Like in SMMs, the system should have an Ising-type magnetic anisotropy, i.e., the spins must preferentially orient in one direction. The main concept of dynamics is the probability of a spin to flip within the chain, taking into account only the nearest neighbors’ interactions, with an Hamiltonian of the type
Ĥ = −
JΣi= 1,Ν−1ŜiŜi+1. The prototype compound is the 1D complex {[Co
II(hfac)
2(NITPhOMe)]}
n (
3) [
40], where hfac is the hexafluoroacetylacetonato(−1) ligand and NITPhOMe is the neutral nitronyl nitroxide, bridging radical 4′-methoxyphenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide. The Ising nature of the chain has been attributed to the presence of Co(II) as this center yields significant anisotropic effects when it is 6-coordinate.
The third class of molecular compounds, of great interest to scientists, consists of multifunctional materials whose study is beyond molecular magnetism. These molecular materials combine two, technologically interesting, electronic properties, e.g., ferromagnetism and superconductivity, or SMM properties and photoluminescence; such properties are very difficult to be included in purely inorganic solids. Examples of crystalline, multifunctional molecular materials include porous magnets [
41], chiral magnets [
42,
43], conducting magnets [
44] and luminescent SMMs [
45,
46], among others.
In the last few years, the interdisciplinary field of molecular magnetism is rapidly shifting to magnetic molecules and materials in physics- and nanotechnology-related fields [
19], such as molecular spintronics, quantum technologies, 2D materials and MOFs [
37,
47,
48,
49,
50,
51]. For example, in the field of quantum technologies, the achievements in the design of molecular spin qubits with long quantum coherence times and in the implementation of quantum operations give hopes for the use of molecular spin qubits in quantum computation.
Closing this introductory section, we would like to emphasize some advantages of the use of nd metal ions (compared to nf ones) in the SMM/SIM research. The magnetic anisotropy of a transition metal center can be more rationally tuned by chemical design. For a given dn configuration, both the sign and the magnitude of D are controlled, mainly by the geometry of the coordination sphere of the nd metal ion, and thus they can be tuned more easily than for a nf metal ion by the chemist. Very large |D| values are found for complexes with low coordination numbers, which are more accessible for the nd metal ions (at least with n = 3) than for the nf metal centers. Such chemical control on the magnetic anisotropy of monometallic SMMs can help scientists to develop more efficient polynuclear molecular magnetic species.
3. Scope and Organization of this Review
As briefly mentioned in
Section 1, in the last 25 years there has been a renaissance in the field of molecular magnetism. The main reason for this was the discovery of the exciting properties and the efforts for improvement of SMMs and SIMs. One of the consequences of these discoveries was an explosive growth of synthetic molecular inorganic and organometallic chemistry. Many groups around the world have been working on the synthesis of SMMs and SIMs with higher effective energy barriers for magnetization reversal (
Ueff) and
TB values, with the Holy Grail in this area being their technological applications. Novel structural types of metal complexes and new, smart synthetic methods have been reported to realize this general goal.
Inorganic and organic ligands are central “players” in this game. Concerning transition-metal ion polynuclear SMMs, the ligands are of various types. The inorganic ligands are mainly the hydroxido, oxido, azido, dicyanamido, cyanato, cyanido and halogenido groups. Simple carboxylato, azolato, deprotonated diol and triol, and thiolato organic ligands are frequently used, but the most employed ones are polydentate ligands involving a combination of functional groups, e.g., alkoxido, phenoxido, carboxylato, oximato, etc. Concerning transition-metal ion monometallic SMMs (SIMs), the ligands should be terminal and the most popular ones are simple heterocycles, phosphines and thioureas, thiolates, phenolates, bis(trimethylsilyl)amide, 2,9-dialkylcarboxylate-1,10-phenantrholines, 6,6′-dialkylcarboxylate-2,2′-bipyridines, chelating Schiff bases and various organometallic-type ligands, including tris(trimethylsilyl)methanide. We should mention here that the
Ueff term is used to indicate thermally induced reversal of the magnetization, the rate of which is dependent on the energy barrier in which the system must surmount to reverse the spin [
30]. Due to tunneling effects,
Ueff is always lower than
U defined in
Section 1.
The general aim of this review is to highlight the use of few (we emphasize the word “few”) inorganic and organic ligands in the chemistry of 3d-, 4d-, and 5d-metal SMMs and SIMs, through selected examples. According to the authors’ opinion (a subjective opinion!), these ligands have contributed much into the development of the transition-metal SMM/SIM area, but simultaneously they are promising and have the potential for exciting achievements in the future. Ligands that have been used in f-metal-containing (both homometallic and heterometallic) SMM and SIM chemistry will not be described.
The review’s content is shaped by a few specific features. First, it is important to specify
what this review is not. It is
not a comprehensive review on the chemistry of transition-metal SMMs and SIMs; there are excellent reviews and books covering this wide topic [
21,
22,
23,
26,
27,
29,
57,
58,
59,
60]. We never considered the idea of being exhaustive; each of the sections could have been on its own a subject of another review. It is
not a survey of recent interesting results; such results can be found in the current literature. Thus, we apologize to the outstanding researchers whose excellent work will not be cited.
Second, the content of the review is chemical and we assume that the readers have a good knowledge of synthetic, structural and physical coordination and organometallic chemistry, as well as of the properties of SMMs/SIMs and the techniques of their study. Structural and magnetic information will be confined to the absolute minimum. To avoid long synthetic discussions, balanced chemical equations (written using molecular and not-ionic formulae) will be used. This, of course, implies that only one reaction occurs in solution (which is certainly not the case, at least in SMM chemistry that involves polynuclear species); however, we do believe that writing chemical equations offers a great help to the reader to understand the processes, better than presenting a long text. We shall try to explain the synthetic philosophy behind the reactions with emphasis on the choice of ligands and metal ion sources. Many of the references for the general information are reviews and book chapters.
Third, the method that will be used to describe the coordination of ligands to transition-metal ions is mainly the “Harris Notation”; occasionally, and in simple cases, the traditional η/μ rotation will be also used. The “Harris Notation” [
61] is an already widely accepted method for the description of the ligands’ binding to metal ions. It is written in the form X.Y
1Y
2Y
3…Y
i, where X is the total number of metal ions bound by the whole ligand, and each Y value refers to the number of metal centers attached to the different donor atoms. The order of Y atoms follows the Cahn–Ingold–Prelog priority rules; hence, for most of the ligands reported in this review, O comes before N. For clarity, the coordination modes of the ligands in most examples of this work will be presented schematically.
A fourth, more personal point, is that this review is a distillation from life-long experience of our group as a synthetic one, preparing molecules and molecular assemblies specially designed to exhibit given physical properties. Few of the ligands that will be discussed reflect our poor knowledge of and minor contributions into the area of the chemistry of SMMs and SIMs with 3d- and 4f-metal ions. Thus, few examples (but not the majority of them) will be work from our group.
Fifth, it is important to realize that the discipline of designing appropriate ligands for efficient transition-metal SMMs and SIMs has reached such a state of maturity that the present attempt cannot give (we are afraid) innovative ideas, but it will be a trip to the great achievements of selected (and not all) groups in the area.
As far as we are aware, this is the first attempt to highlight in a review the great influence of ligands on SMM and SIM properties. The topic has been partly covered in many of the books [
21,
22], chapters in books [
55] and reviews [
23,
26,
27,
57,
58,
59,
60] available dealing with the chemistry, physics, properties and potential applications of molecular nanomagnets.
4. The Azide Ion: An Evergreen “Tree” in the Chemistry of Transition-Metal SMMs
The azide ion (N
3−) is the conjugate base of hydrazoic acid. Aqueous solutions of HN
3 were first prepared by T. Curties in 1890, Equation (3). Such solutions are weakly acidic (pK
a = 4.75).
The azido ligand is very popular in transition-metal chemistry and especially for the synthesis of coordination clusters and coordination polymers [
62,
63,
64]. In Coordination Chemistry, it is found either as a terminal ligand (η
1) or as a bridging one. The to-date crystallographically established coordination modes of the bridging azido ligand are illustrated in
Figure 3.
The bridging azido ligand has been one of the most investigated ligands in Magnetochemistry and Molecular Magnetism [
63]. The variety of its bridging modes and its ability to propagate exchange interactions has led to compounds with several kinds of magnetic behaviors, such as antiferromagnetism, ferrimagnetism, ferromagnetism, canted weak ferromagnetism, spin-flop, and SMM and SCM properties [
63]. End-to-end (EE) azido groups generally propagate antiferromagnetic interactions between paramagnetic metal ions, whereas end-on (EO) N
3− ions are generally ferromagnetic couplers; exceptions of this general rules are known [
63]. In addition to the coordination mode, other structural and electronic parameters (bridging and dihedral angles, bond lengths, orthogonality of magnetic orbitals, spin polarization, delocalization of unpaired electrons, et al.) play an important role in determining the sign and strength of the magnetic coupling. The ability of N
3− to promote ferromagnetic exchange interaction has been utilized in the SMM chemistry of 3d-metal ions [
62,
63,
64].
We should mention here that an excellent experimental and theoretical study by Sarkar, Neese, Meyer and coworkers [
65] has opened the doors for the use of the terminal azido group in 3d-metal SIM chemistry. Contrary to previous suggestions, it was shown that the N
3− ligand behaves as a strong σ and π donor. Magnetostructural correlations have revealed a remarkable increase in the negative
D value with shortening of the axial Co
II–N
azido bond lengths, i.e., with increasing Lewis basicity.
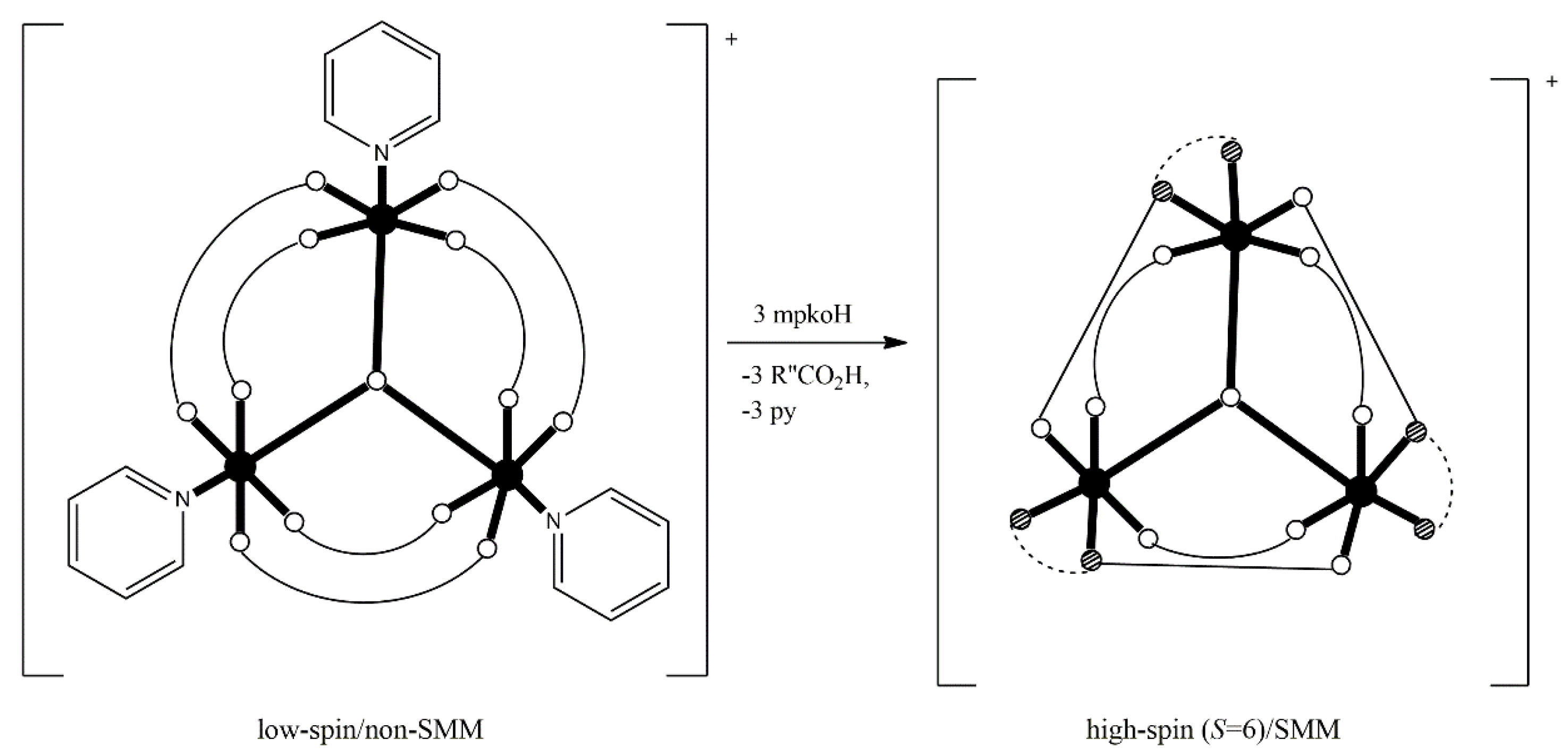
Returning to SMM chemistry, our group, in collaboration with the group of Escuer at Barcelona, have developed a general synthetic strategy for the remarkable increase of the ground-state spin in coordination clusters, which often “switches on” SMM behavior [
66,
67,
68,
69]. The strategy is based on the substitution of bridging hydroxido or alkoxido groups, which most often propagate antiferromagnetic exchange interactions, in pre-formed coordination clusters by EO azido groups which propagate ferromagnetic interactions. The core changes, but the nuclearity does not. The incoming azido groups (always EO) introduce ferromagnetic components in the superexchange scheme of the molecule and, as a consequence, the ground-state spin increases significantly, sometimes inducing SMM behavior.
The reaction between the pre-formed cluster [Fe
II9(OH)
2(O
2CMe)
8{(py)
2C(O)
2}
4] (
5) and a slight excess of NaN
3 in refluxing MeCN under N
2 gives the corresponding azido cluster [Fe
II9(N
3)
2(O
2CMe)
8{(py)
2C(O)
2}
4] (
6) in ~40% yield, Equation (4); (py)
2C(O)
22− is the dianion of the
gem-diol form of di-2-pyridyl ketone, (py)
2CO [
68,
69]. The reactant and the product have similar molecular structures, the only difference being the presence of two 4.40 azido groups in the latter instead of two 4.4 hydroxido groups in the former. The nine Fe
II atoms in
6 adopt the topology of two square pyramids sharing a common apex. Bridging within each square base is achieved through four syn, syn 2.11 MeCO
2− ligands, four alkoxido oxygen atoms from the four 5.3311 (py)
2C(O)
22− ligands (
A in
Figure 4) and one extremely rare 4.40 azido group (
Figure 3). Each alkoxido oxygen is μ
3 bridging connecting two metal centers from a square base to the central Fe
II atom; the latter is thus 8-coordinate. The core is {Fe
II9(4.40-N
3)
2(μ
3-OR)
8}
8+,
Figure 5. The two square bases have a slightly staggered conformation which results in a square antiprismatic coordination geometry about the central Fe
II atom, the chromophore being {Fe
IIO
8}. Dc magnetic susceptibility studies in the 2–300 K temperature range indicate that the substitution of the 4.4 OH
−s in
5 by the 4.40 N
3−s in
6 induces ferromagnetic coupling in the latter; its ground spin state is not well isolated from low-lying excited states and it cannot be accurately determined. Compound
6 is EPR silent at the X-band frequency at 4.2 K, but it is an SMM with a
Ueff value of 29(1) cm
−1. The slow magnetic relaxation is also evident using zero-field
57Fe Mössbauer spectroscopy.
Almost all the azido-bridged transition-metal clusters and SMMs contain chelating or bridging organic ligands. The presence of bridging organic ligands is usually a disadvantage because it introduces antiferromagnetic components in the superexchange scheme, thus decreasing the effect of the ferromagnetic EO azido groups. The group of Stamatatos have recently developed [
70,
71,
72] a novel strategy for the synthesis of 3d-metal, EO azido-bridged coordination clusters with high-spin
S values and SMM properties by avoiding the presence of any organic chelating/bridging ligand. The azido groups in the cores of the clusters are exclusively EO, which ensures the presence of ferromagnetic exchange interactions between the metal spin carriers and therefore the attainment of the maximum
S value. In the presence of magnetic anisotropy, induced by the choice of the appropriate metal ion, some of the clusters also exhibit remarkable SMM properties. The peripheral ligation around the metallic core is completed by terminal, volatile MeCN ligands, whose solvation/de-solvation effects sometimes lead to interesting magnetic phenomena. The key reagent for this chemistry is Me
3SiN
3. This has some remarkable differences compared to its all-inorganic analogue, which is almost 100% used in metal–azide chemistry. Firstly, Me
3SiN
3 is more soluble in organic solvents than NaN
3, and this allows reactions to be performed in a variety of such solvents. Secondly, Me
3SiN
3 can abstract OH
− ions from the reaction media and thus bridging hydroxide ligands are not incorporated in the products; this allows the dominance of the N
3− ions in solution which can act as ligands without competition from the OH
− species. And third, Me
3Si
+ cannot coordinate and Na
+ ions are not incorporated in the clusters, which is often the case when NaN
3 is used as the azide source. Representative examples of this strategy are briefly mentioned below.
The 1:1:4 reaction between Fe
II(ClO
4)
2∙6H
2O, Et
3N and Me
3SiN
3 in MeCN under N
2 gives a dark red solution from which orange crystals of [Fe
II7(N
3)
12(MeCN)
12](ClO
4)
2∙4MeCN (
7∙4MeCN) can be isolated in a ~55% yield, Equation (5). The crystals were treated in two ways for magnetic and spectroscopic studies [
70,
71]. A portion of the crystalline material was immediately transferred and sealed in an NMR tube representing the structurally characterized sample. The other portion was collected by filtration and dried under N
2 for 3 h; its analytical data corresponded to the formula {Fe
II7(N
3)
12(MeCN)
2(ClO
4)
2} (
7a). The IR spectra of
7∙4MeCN and
7a indicate the presence of the EO azido group, with the band due to the
νas(N
3) mode appearing at ~2100 cm
−1. The almost complete de-solvation of
7∙4MeCN to give
7a is evidenced by the nearly complete disappearance of the IR bands due to the
ν(C≡N) mode of the MeCN molecules from the spectrum of the latter (at 2310 and 2279 cm
−1 in the spectrum of the former).
The heptanuclear cation that is present in the crystal structure of
7∙4MeCN (
Figure 6) contains a nearly ideal planar hexagon of metal ions centered on the seventh, central Fe
II atom [
71]. The central Fe
II atom is a crystallographic inversion center; the {Fe
II7} disk-like cation possesses virtual S
6 symmetry. The seven octahedral Fe
II atoms are held together through six 2.20 and six 3.30 EO azido groups. The six 3.30 azides connect the outer {Fe
II6} hexagon with the central metal ion, while the six 2.20 azides bridge exclusively the outer Fe
II centers. Peripheral ligation is completed by twelve MeCN molecules, two on each of the outer metal ions. The {Fe
II7(3.30-N
3)
6(2.20-N
3)
6}
2+ core can also be described as consisting of six {Fe
II3(N
3)
4} defective cubane units, each double face-sharing; a vertex of each cubane unit is shared with the common vertex of the six cubanes which is the central Fe
II atom. The metal–nitrogen bond lengths are indicative of high-spin (t
2g)
4(e
g)
2 Fe
II atoms with N-ligation. The intramolecular Fe
II∙∙∙Fe
II distances are ~3.35 Å, while the Fe
II–N–Fe
II angles span the range 95.5–105°. The intermolecular Fe
II∙∙∙Fe
II distances are large (>8 Å) due to the packing of the heptanuclear cations and to the presence of the coordinated MeCN molecules.
Both the as-synthesized (
7∙4MeCN) and dried (
7a) forms of the complex were magnetically studied [
70,
71]. Dc magnetic susceptibility studies reveal the maximum possible ground-state spin (
S = 14). Both forms are SMMs; however, their ac magnetic dynamics are different, revealing a “Janus”-faced SMM behavior for the pristine and dried samples which have been attributed to solvation/de-solvation effects from the coordinated solvent molecules. Sample
7a exhibits two individual relaxation processes, which are both thermally assisted; the 2.7–5.0 K process is characterized by a
Ueff value of 30.5(1) cm
−1, and the 1.8–2.6 K process by a
Ueff value of 15.3(2) cm
−1. Data are shown in
Figure 7. In contrast, the as-synthesized (pristine) sample
7∙4MeCN exhibits only one relaxation process below ~3 K with a
Ueff value of 10.0(2) cm
−1. The different number of relaxation processes and the different values of effective energy barriers for magnetization reversal can be rationalized in terms of the differences in intermolecular, i.e., intercationic, interactions and the different molecular anisotropies arising from different crystal fields around the peripheral Fe
II atoms [
71].
Complexes [Co
7(N
3)
12(MeCN)
12](ClO
4)
2 (
8) and [Ni
7(N
3)
12(MeCN)
12](ClO
4)
2 (
9) were prepared by a reaction similar to that used for
7∙4MeCN, simply replacing the metal perchlorate starting material [
72]. The crystal structures of
8 and
9 (these complexes are isomorphous) do not contain solvent molecules in the lattice. The clusters have a similar molecular structure to its {Fe
II7} analogue. The crystals of
8 and
9 are stable at room temperature and no degradation is observed after 24 h exposure to the normal laboratory atmosphere; the static and dynamic properties of their wet- and “dried”-forms are identical for each complex. As expected, both clusters are strongly ferromagnetically coupled. The Ni(II) cluster (
S = 7) has a negligible magnetoanisotropy and, consequently, it does not exhibit out-of-phase ac magnetic susceptibility signals, i.e., it is not a SMM, in either the absence or the presence of an external dc field. The Co(II) cluster exhibits SMM properties under the application of a weak external dc field of 0.1 T with a
Ueff value of 19.6(1) cm
−1 [
72]. The application of weak dc fields during the dynamic susceptibility studies helps to suppress QTM, which is otherwise strong for systems in low-symmetry crystal environments. This experimental practice is very helpful in elucidating the mechanisms operating in magnetization relaxation processes. However, the researchers should be cautious when interpreting ac susceptibility results in the presence of external dc fields; an excellent description of such a cautionary note is provided in reference [
27].
The great utility of the above mentioned synthetic approach, which can potentially stimulate the research in transition-metal azido chemistry to meet new directions, is demonstrated by the 2-nm-sized spherical cluster [Mn
29O
24(N
3)
10(dma)
28] (
10) [
73], where dma is the 3,3-dimethylacrylate(−1) ligand. The cluster contains a {Mn
IIMn
III28(4.4-O)
8(3.3-O)
16(4.40-N
3)
2(2.20-N
3)
8}
28+ core, and has an
S = 9/2 ground state. Despite the appreciable number of EO azido groups, the complex is not an SMM due to the simultaneous presence of bridging oxido and carboxylato groups, which promote antiferromagnetic exchange interactions and “force” the Mn–N
azido–Mn angles to be rather large (average 111.4°) presaging antiferromagnetic interactions between the respective azido-bridged metal ions.
5. Cyanido-Directed Assembly of Transition-Metal SMMs
Many transition-metal SMMs contain hydroxido (OH
−), alkoxido-type (RO
−) or oxido (O
2−) bridging groups. The problem is that the oxygen atom can bridge two or more (up to six with the O
2− ligand) metal centers, with a wide variety of M–O–M angles. Since the pairwise magnetic exchange interactions (which should be ferromagnetic and strong for an efficient SMM) are very sensitive to the bridging angles (and more generally to local geometry), chemists cannot often predict the magnetic properties of a complex structure. As a consequence, the search for new metal–hydroxido/alkoxido/oxido SMMs remains a rather serendipitous effort. The “good news” is that there is an alternative small-sized inorganic bridging ligand which might overcome the aforementioned difficulty. This is the cyanide (CN
−) group. Given that Prussian Blue, Fe
III4[Fe
II(CN)
6]
3∙
xH
2O (
x = 14–16), the first coordination complex and the first molecule-based magnetic solid was discovered in 1704 by the painter Diesbach in Berlin, it is amazing that after more than 300 years the CN
− ion is in the forefront of coordination chemistry and molecular magnetism [
74]. Perhaps, Andreas Ludi, who called Prussian Blue “An Inorganic Evergreen” in an article written in 1981, was more visionary when he gave this nickname [
75].
The preference of CN
− for binding just two metal sites, one at each end, leading to a linear bridging arrangement is well established in inorganic chemistry. Thus, solution assembly reactions can be designed with the expectation that the product will possess linear M–CN–M’ groups, thus providing synthetic chemists with a degree of synthetic and structural control [
58]. In addition, because of the linear bridging arrangement, there is a satisfactory level of predictability in the expected nature of the magnetic exchange coupling between octahedral M and M’ spin carriers. Unpaired spin density from orthogonal metal-based orbitals (t
2g + e
g) will leak over into orthogonal CN
−-based orbitals, leading to ferromagnetic exchange via Hund’s rule. On the contrary, the unpaired spin density from metal-based orbitals of compatible symmetry (t
2g + t
2g or e
g + e
g) will leak over into the same cyanide-based orbitals, leading to antiferromagnetic exchange via Pauli exclusion principle. The antiferromagnetic exchange interactions are generally stronger and tend to dominate the superexchange in a competitive coupling scheme. Such predictions are useful in the design of cyanido-bridged clusters with high values of the ground-state spin [
21,
55,
58].
Metal–cyanido chemistry can rather easily lead to molecules with high-spin ground states [
21,
58]. The difficult problem is to introduce axial magnetic anisotropy in these systems, i.e., negative
D. The incorporation of metal centers that have a large single-ion anisotropy is a good first step in obtaining large
D values in a cluster. The anisotropy of an individual metal ion is determined by the coupling between its spin angular momentum and its orbital angular momentum. Thus, metal ions with orbitally degenerate ground states are expected to have a large zero-field splitting and would be appropriate for incorporation in SMMs. This is, for example, the case in
1 (
Figure 1) where the outer Mn
III atoms, with a t
2g3e
g1 configuration contribute much to the anisotropy barrier. Thus, there are two approaches for instilling magnetic anisotropy in metal–cyanido clusters. The first one is to utilize metal ions already known to give hydroxido-, alkoxido- and/or oxido-bridged 3d-metal SMMs, e.g., V
III, high-spin Mn
III, high-spin Co
II, etc. Such high-spin configurations are obtained easily with O-based donors; however, low-spin configurations often occur in metal–cyanido clusters and certain of these (with orbital degeneracy) may be particularly effective. The second approach is to utilize 4d- and 5d-metal ions. Because of the relativistic nature of the spin-orbit coupling phenomenon (which is a source of magnetic anisotropy), this will generally result in significant increase of single-ion anisotropy for heavier transition-metal ions. For example, replacement the Cr
III atoms in known cyanide clusters with Mo
III atoms can lead to clusters with enhanced magnetic anisotropy while preserving the ground-state spin [
58]. Of particular importance in this context is that theory has predicted that single-ion anisotropy originating from spin-orbit coupling will give SMMs with higher
TB values [
76].
Based on the facts mentioned above, the main method for synthesizing cyanido-based homo- and, especially, heterometallic transition-metal SMMs is a building-block approach, often called modular process or “complexes as ligands and complexes as metal ions” strategy. A typical cluster preparation involves two building units. One bears one or more coordination sites occupied by labile (loosely coordinated) solvent molecules (the “metal ion”) and the other is a complex that possesses one or more terminal cyanido ligands (the “metalloligand”) [
21,
55,
58,
77,
78,
79,
80,
81,
82,
83,
84,
85]. In solution, the nucleophilic “free” nitrogen atoms of the terminal cyanido ligands displace the labile solvent molecules, leading to dinuclear or polynuclear assemblies. An important synthetic parameter is the nature and denticity of the capping (chelating) ligands for either of the two building units (precursors); these characteristics often control the nuclearity of the product. The “metal ion” in this building-block approach can also be a simple metal “salt” without a capping ligand and/or labile solvent molecules. In addition to this structural control, there are available qualitative rules for predicting the nature and the strength of the magnetic exchange interaction between the cyanido-bridged metal spin carriers established via detailed studies on hundreds of Prussian Blue analogues [
15,
21,
58,
86]. This fruitful combination of structural and magnetic predictability has led to the rational design, synthesis and study of many cyanido-bridged SMMs.
A significant advantage of transition metal–cyanido cluster chemistry is that once a stable new structural motif is identified, the researcher can be confident that she/he can replace some metal ions with certain other metal ions in the structure [
58].
After this, rather long, introduction we proceed with few representative examples. The metalloligand (Et
4N)[Re
II(CN)
3(triphos)], where triphos is the bulky tridentate phosphine 1,1,1-tris(diphenylphosphinomethyl)ethane (
Figure 8a), was prepared by Dunbar’s group. Direct reaction of [(triphos)Re
II(μ-Cl)
3Re
II(triphos)]Cl with 6 equivs of (Et
4N)CN in MeCN leads to homolytic scission of the {Re
II≡Re
II}
4+ unit to give the bright yellow precursor metalloligand, Equation (6), in ~35% yield [
87]. The coordination geometry of Re
II in the mononuclear anion is
fac pseudo-octahedral; three coordination sites are occupied by the C-bonded cyanido ligands, while the other three sites are filled by the P atoms of the tridentate chelating triphos ligand. Mononuclear 17-electron complexes of Re(II) are attractive in molecular magnetism due to strong spin-orbit coupling effects (
λ = 2000–3000 cm
−1) arising from the low-spin 5d
5 configuration. The 1:1 reaction between (Et
4N)[Re
II(CN)
3(triphos)] and anhydrous MnCl
2 in Me
2CO/MeCN under reflux gives the orange-red cluster [Mn
II4Re
II4Cl
4(CN)
12(triphos)
4] (
11), Equation (7). The octanuclear molecule has an approximate cubic topology (
Figure 8b) [
78,
79] with alternating octahedral Re
II and tetrahedral Mn
II concerns. The edges of the cube are spanned by the 12 bridging cyanido groups (Re
II–CN–Mn
II) that link the metal ions. The coordination geometry around the Re
II atoms is similar to that of the mononuclear starting metalloligand, i.e., distorted octahedral with the triphos ligand behaving as a facially capping, tridentate chelating ligand with the carbon end of the three cyanido ligands completing the coordination sphere. The Mn
II centers adopt a distorted tetrahedral geometry, the donor atoms for each metal ion being three coordinated nitrogen atoms from bridging cyanido groups and a fourth terminal chlorido ligand extending out of the cube. It is obvious that the steric bulk of the triphos ligands enforces the distorted tetrahedral coordination environments at each of the 3d-metal sites. The structurally similar cubic complexes [Mn
II4Re
II4I
4(CN)
12(triphos)
4] (
12a) and [M
II4Re
II4Cl
4(CN)
12(triphos)
4] (M = Fe,
12b; M = Co,
12c; M = Ni,
12d; M = Zn,
12e) have also been prepared through analogous reactions [
79]. Variable-temperature dc magnetic susceptibility data for
11 indicate antiferromagnetic coupling between the “
S = 1/2” Re
II and
S = 5/2 Mn
II atoms. The complex is a weak SMM with a
Ueff value of ~9 cm
−1. Micro-SQUID temperature-dependent scans reveal hysteretic behavior for the cluster. The data also show a prominent step at zero field, resulting from fast QTM. The step becomes temperature-independent below 0.2 K but remains sweep rate-dependent, suggesting a ground-state resonant tunneling process at
H = 0 [
79].
Staying at Re, an interesting metalloligand is [Re
IV(CN)
7]
3−, which was prepared by Long’s group through a simple ligand metathesis reaction [
88], Equation (8). This anion has a low-spin 5d
3 pentagonal bipyramidal geometry. The complex has a strong magnetic anisotropy which can be attributed to a combination of its large spin-orbit coupling associated with the heavy rhenium ion and the unquenched orbital angular momentum of its
2E
1’ electronic ground state. Thus, the incorporation of [Re
IV(CN)
7]
3− into a high-spin coordination cluster would “transfer” magnetic anisotropy to the product, possibly leading to slow magnetic relaxation. Complex [Mn
II(PY5Me
2)(MeCN)](PF
6)
2 is an ideal “metal ion” for a building-block reaction for three reasons: (i) The Mn
II ion has 5 unpaired electrons; (ii) the six-coordinate cation possesses a labile MeCN molecule; and (iii) the potentially pentadentate chelating ligand 2,6-bis(1,1-bis(2-pyridyl)ethyl)pyridine ligand (PY5Me
2,
Figure 9a) is an efficient capping moiety which can ensure the formation of star-like clusters that are magnetically isolated. Reaction of (Bu
n4N)
3[Re
IV(CN)
7] with 4 equivs of [Mn
II(PY5Me
2)(MeCN)](PF
6)
2 in MeCN at −40 °C produces a blue solution from which the blue, temperature-sensitive cluster [Mn
II4Re
IV(CN)
7(PY5Me
2)
4](PF
6)
5 (
13) is isolated in a good yield, Equation (9). If the same reaction is performed at room temperature, an immediate color change from blue to green and then to yellow is observed affording complex [Mn
II4Re
III(CN)
7(PY5Me
2)
4](PF
6)
4 (
13a); this complex is formed via a spontaneous reduction of Re
IV (
S = 1/2) to Re
III (
S = 0) within the cluster [
80].
X-ray analysis on single crystals of
13 reveals the presence of a four-point star-like topology for the cation of the complex, as shown in
Figure 9. The [Re
IV(CN)
7]
3− unit is at the center of the star and is bridged through cyanido groups to four {Mn
II(PY5Me
2)}
2+ pendant units; three cyanido ligands remain terminal at the central Re
IV atom. The coordination polyhedron of the 5d-metal ion is close to that of an ideal pentagonal bipyramid, with an essential liner axial C
ax–Re
IV–C
ax angle of 179.9°. The arrangement of the four 3d-metal ions can be described as a square, with two of the Mn
II atoms binding axial cyanido groups of Re
IV and the other two binding non-neighboring equatorial cyanido groups of Re
IV. The magnetic exchange interactions between the central Re
IV atom (
S = 1/2) and the surrounding Mn
II centers (
S = 5/2 each) are ferromagnetic, resulting in a high-spin ground state (most probably 21/2). The high-spin ground state of the cluster, combined with the negative
D value of −0.44 cm
−1 gives SMM properties in
13 with an effective relaxation barrier of
Ueff = 33 cm
−1 [
80]. Analogous reaction schemes lead to structurally similar clusters [M
II4Re
IV(CN)
7(PY5Me
2)
4](PF
6)
5 (M = Ni,
13b; M = Cu,
13c), which are also SMMs with
Ueff values of ~17 and ~8 cm
−1, respectively [
81]; the
D values are −0.93 cm
−1 (
13b) and −1.33 cm
−1 (
13c), the corresponding
S values being 9/2 and 5/2.
Today the cyanido ligand is one of the most popular ligands in SMM chemistry [
21,
85], because of the variety of metalloligands that are available. For example, octacyanidometallates are unique building blocks that are useful in the construction of various types of magnetic clusters with topologies ranging from square, trigonal bipyramidal, octahedral, to pentadecanuclear six-capped-body-centered cubes and even larger molecules, some of which are SMMs [
85].
6. Tris(trimethylsilyl)methanide, an Old Organometallic “Friend” Joins the Chemistry of 3d-Metal SIMs
Several groups around the world re-investigate the magnetic anisotropies of complexes based on transition-metal ions in a general effort to find magnetic alternatives to the f-block metal ions. As it has already been mentioned in
Section 1, mononuclear transition metal–ion complexes are not good candidates for large
Ueff values because of their smaller magnetic moments and, for the 3d-metal ions, lower spin-orbit coupling constants. Also, the larger ligand-field splitting energies of the d orbitals lower the orbital contributions to the magnetic properties required to develop significant magnetic anisotropy. There are two manifestations of this effect: (i) The first-order orbital angular momentum can be quenched as the result of a Jahn–Teller distortion; and (ii) the second-order contribution to the magnetic anisotropy, i.e., the zero-field splitting, is becoming very small due to the large energy separation between ground and excited electronic states, which reduces the degree of mixing. These ligand-field effects can become much less important by synthesizing transition metal–ion complexes with very low coordination numbers [
27]. In this way, the ligand-field splitting energies of the d orbitals fall within a narrow range, similar to the energies of the 4f orbitals in Ln(III) complexes. A linear two-coordinate geometry at the metal center is ideal for suppressing ligand-field effects in 3d-metal complexes and generating a large anisotropy barrier. For a strictly linear coordination geometry, with a local D
∞h symmetry at the metal ion, the energies of the d orbitals are split as (d
xy, d
x2−y2) < (d
xz, d
yz) < d
z2 (these relative energies do not consider the possibility of s-d
z2 mixing, which is known to lower the energy of d
z2 below that of the d
xy, d
yz pair), with the d
z2 and (d
xz, d
yz) orbital energies being destabilized by σ- and π-metal–ligand interactions, respectively; on the contrary, the (d
xy, d
x2−y2) orbitals have δ symmetry and are thus not participating in bonding with the ligands’ orbitals. A high-spin 3d
6 electron configuration is expected to give a large first-order contribution to orbital angular momentum that will not be quenched through a Jahn–Teller distortion; this maximizes the magnetic moment and the two donor atoms define an axis for its easy alignment [
89].
One of the ligands that has played an important role in the chemistry of two-coordinate 3d-metal SIMs is the tris(trimethylsilyl)methanide,
−C(SiMe
3)
3. The neutral compound HC(SiMe
3)
3 and its conjugate base have their own history in organic [
90,
91] and organometallic [
92,
93] chemistry, respectively. The tris(trimethylsilyl)methanide (‘trisyl’) anion has been used as a very bulky ligand in main group and Ln(III) chemistry. The advantages of this group are numerous: (a) Due to its bulkiness, it can lead to very low coordination numbers and stabilize coordinatively unsaturated transition-metal complexes which often exhibit unusual structural features or exciting reactivity; (b) its steric bulk-which is estimated to be similar to that of Cp* anion or P(
tBu)
3-confers kinetic stability to complexes; and (c) it lacks α-and β-hydrogens prohibiting undesired reactivity. In a sense, it may be viewed as combining the electronic features of a methyl anion and the steric requirements of the Cp* anion [
93].
Complex [Fe
II{C(SiMe
3)
3}
2] (
14) was synthesized [
93,
94] by the reaction illustrated in Equation (10). Li{C(SiMe
3)
3}∙2THF can be prepared by the metallation of tris(trimethylsilyl)methane with MeLi in THF/Et
2O under reflux [
92]. The two-coordinate Fe
II center sits on a crystallographic inversion center, resulting in a strictly linear geometry (C–Fe
II–C = 180.0°). Variable-temperature dc magnetic susceptibility data indicate a high-spin 3d
6 Fe
II center (t
2g4e
g2). Low-temperature magnetization data show a saturation value of 3.24 B.M.; this value is lower than 4 B.M. that would be expected for a spin-only
S = 2 center. The results from the dc susceptibility and magnetization data indicate that
14 has a highly anisotropic magnetic moment. Both data sets could not be fit, suggesting that the magnetic anisotropy of the molecule is not due to spin-only phenomena [
94]. Ac susceptibility data reveal slow magnetic relaxation under an external dc field. Arrhenius plots for the linear complex were fit by employing a sum of tunneling, direct, Raman and Orbach relaxations, resulting in a
Ueff value of ~146 cm
−1. Theoretical calculations (CASSCF/NEVPT2 on the crystal structure) were performed to explore the influence of deviation from rigorous D
∞h geometry on the splittings of the 3d orbitals and the electronic state energies. The calculations suggest that the ligand field asymmetry quenches the orbital angular momentum of
14, but finally spin-orbit coupling is strong enough to compensate and regenerate the orbital moment. The non-observation of a single Arrhenius behavior has been attributed to a combination of the asymmetry of the ligand field and the influence of vibronic coupling.
The observation of slow magnetic relaxation in
14, and related mononuclear two-coordinate Fe(II) complexes, requires the application of an applied dc field which suppress fast magnetization reversal through QTM; the latter is very efficient for such systems because of the small non-Kramers
S = 2 ground state prohibiting slow relaxation in zero magnetic field [
27]. An alternative way to minimize tunneling of the magnetic moment, caused by mixing of the ground-state ±
MS levels, is employment of half-integer spin systems, according to the Kramers’ theorem [
95]. Such systems should not require the application of a dc field to display slow magnetic relaxation. The group of Long reported the structurally interesting and magnetically impressive two-coordinate SIM [K(crypt-222)][Fe
I{C(SiMe
3)
3}
2] (
15), which possesses a
S = 3/2 ground state [
96]; crypt is the ligand 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8,8,8]-hexacosane, which chelates the K
+ ion though its six ether oxygen atoms. The synthesis of
15 was achieved by the one-electron reduction of
14 using KC
8 as reductant, Equation (11). The possibility of the one-electron reduction of the Fe(II) complex had been recognized by cyclic voltammetry studies in difluorobenzene, which show a reversible process corresponding to the [Fe{C(SiMe
3)
3}
2]
0/−1 couple. The product is obtained in yields of ~50% and has a bright yellow-green color. Single-crystal X-ray crystallography reveals a linear geometry around Fe
I (
Figure 10a), the C–Fe
I–C bond angle being 179.2°. The SiMe
3 groups are eclipsed, contrary to the structure of
14, in which they are staggered. Complex
15 not only shows, as expected, one of the highest
Ueff values [226(4) cm
−1] yet reported for SMMs and SIMs that contain transition-metal ions, magnetic hysteresis below 29 K and a
TB value of 4.5 K, but also has improved permanency in zero field [
96,
97]; the
Ueff value starts to approach values that we meet in Ln(III)-based systems. Quantum-chemical and spectroscopic studies suggest an electronic structure (
Figure 10b), which would not be expected by most coordination chemists. The theoreticians in the research team provided strong evidence that strong 4s-3d
z2 mixing stabilizes the 3d
z2 orbital. The result is supported by advanced spectroscopic characterization, and the calculated orbital degeneracies explain well the unquenched orbital moment. The
Ueff value is close to the calculated energy gap between the ground
MJ = 7/2 pair and the first excited
MJ = 5/2 doublet, suggesting that the relaxation occurs via this latter state. Excellent
57Fe-Mössbauer studies of
14 and
15 under zero applied dc field between 295 and 5 K yield their magnetization dynamics on a significantly faster time scale (the lifetime of the measurement is 10
−8 to 10
−9 s) than it is possible with ac magnetometry. From the modeling of the Mössbauer profiles, Arrhenius plots between 295 and 5 K were obtained for the two complexes [
98]. The high-temperature regimes suggest Orbach relaxation with
Ueff values of 178(9) (
14) and 246(3) (
15) cm
−1, in good agreement with the values obtained from magnetism. In
15, two distinct high-temperature regimes of magnetic relaxation are observed with mechanisms corresponding to two distinct single-excitation Orbach processes within the ground-state spin–orbit coupled manifold of the Fe
I atom.
It has become clear from the preceding information that orbital angular momentum
L gives rise to magnetic anisotropy, an essential property for efficient SIMs. Unquenched
L arises from an odd number of electrons in degenerate orbitals; this is observed only for free ions and in f-element complexes. In transition-metal complexes, however, the ligand field removes any orbital degeneracy, leading to practically zero
L. Any magnetic anisotropy in such complexes is a weak effect arising from mixing of the electronic ground state and excited states induced by spin–orbit coupling. The maximum value
L for a transition metal is 3 and, at first glance, it seems impossible. An
L = 3 ground state in a linear complex requires two sets of degenerate orbitals, (d
x2−y2, d
xy) with m
l = ±2 and (d
xz, d
yz) with m
l = ±1, and an odd number of electrons in each. Such a system would imply a non-Aufbau configuration, wherein the electrons do not fill the d orbitals in the usual manner from lowest to highest energy, and likely exhibit a large magnetic anisotropy. Having as scientific arsenal: (A) The previous characterization of
15 with unquenched orbital angular momentum, large mangetic anisotropy and non-influence by Jahn–Teller distortions that would otherwise remove orbital degeneracy. (B) Experiments which have shown that Co atoms, deposited on a MgO surface under vacuum (referred to as adatoms) and adopting a coordination number of 1, have a
J = 9/2 (
S = 3/2,
L = 3) ground state giving rise to near-maximal magnetic anisotropy [
99]; and (C) Calculations on the hypothetical complex [Co
II{C(SiMe
3)
3}
2] which have shown an
L = 3 ground state arising from a non-Aufbau (d
x2−y2, d
xy)
3, (d
xz, d
yz)
3(d
z2)
1 filling of the 3d orbitals and further predicting a gap of ~455 cm
−1 between ground and first excited
MJ states [
100], a mutli-national research team led by Long, Neese and van Slageren set out efforts to synthesize and characterize [Co
II{C(SiMe
3)
3}
2], i.e., the 3d
7 analogue of
14.
The strongly reducing nature of the carbanion ligand
−C(SiMe
3)
3 hinders isolation of the desired compound [
101]. Metathesis reactions of {C(SiMe)
3}
− salts and CoX
2 (X = Cl, Br, I) gave only amorphous solids that could not be characterized. However, lowering the basicity of the central carbanion through the introduction of electron-withdrawing aryloxide groups provided access to the dialkyl complex [Co
II{C(SiMe
2ONaph)
3}
2] (
16), where Naph is the naphthyl group. The synthetic process is illustrated in
Figure 11. The Zn(II) congener, [Zn{C(SiMe
2ONaph)
3}
2] (
16a) was obtained in an analogous manner. Using the same reaction conditions with a mixture of ZnBr
2 and CoBr
2∙THF (molar ratio ~900:1) enabled the preparation of the magnetically dilute sample Co
0.02Zn
0.98{C(SiMe
2ONaph)
3}
2 [
101].
Complexes
16 and
16a are isomorphous and feature a linear C–M
II–C axis imposed by the S
6 site symmetry. The staggered orientation of the ligands facilitates close sp
3–CH∙∙∙π and sp
2–CH∙∙∙π contacts (2.69 and 2.82 Å, respectively). This indicates that interligand interactions contribute into the stabilization of the structures [
101]. Ab initio calculations on
16 predict a ground state with
S = 3/2,
L = 3 and
J = 9/2 which arises from the non-Aufbau electron configuration (d
x2−y2, d
xy)
3(d
xz, d
yz)
3(d
z2)
1,
Figure 12; this deviates from the expected Aufbau filling of (d
x2−y2, d
xy)
4(d
xz, d
yz)
2(d
z2)
1. This deviation can be explained in terms of the competing effects of ligand-field stabilization and interelectron repulsion. As for Ln(III) complexes, the ligand field is weak so that interelectron repulsion and spin–orbit coupling determine the electronic ground state. Dc magnetic susceptibility results reveal a well-isolated
MJ = ±9/2 ground state. Variable-field far-infrared spectra suggest a magnetically active excited state at ~450 cm
−1 that, together with variable-temperature ac susceptometry and theoretical calculations, has assigned to the
MJ = ± 7/2 state. A d-orbital filling scheme with equally occupied of (d
x2−y2, d
xy) and (d
xz, d
yz) orbital sets is also indicated by modeling of experimental charge density maps. The
Ueff barrier of ~450 cm
−1 determined for
16 is the largest reported to date for a transition-metal SMM or SIM. As a consequence of its large orbital angular momentum, the magnetically dilute sample Co
0.02Zn
0.98{C(SiMe
2ONaph)
3}
2 exhibits a coercive field of 600 Oe at 1.8 K. Although its magnetic properties mainly pertain at very low temperature, the synthesis, structure and properties of
16 provide scientists with a valuable, general design principle [
101].
7. Softer Ligands for More Efficient SIMs
The main advantage of transition-metal SIM chemistry is that the ground-state spin of the mononuclear molecule is fixed and
D is the only parameter that affects
Ueff. As we have seen, the orbital angular momentum is the main factor that dictates the magnitude of
D; the former depends on the oxidation state and the coordination number/geometry of the 3d-metal ion (for first-row transition metals). Thus, scientists can fine-tune the electronic structure of the complex by playing with the oxidation state and the geometry of the metal ion. The classical approach involves the lowering of the coordination number which provides the system with a large orbital angular momentum resulting in high
Ueff values [
94,
96,
101,
102]. In addition to this approach, several efforts have been performed to alter the spin Hamiltonian parameters of various metal ions by other methods [
103]. Slow magnetization relaxation is normally not observed in integer-spin systems, even under application of external dc magnetic fields, because of underbarrier tunneling mechanisms. As a consequence of this, the interest has focused on non-integer spin systems for a better design of SIMs. Cobalt is a good candidate for efficient SIMs due to strong first-order spin-orbit coupling displayed by the metal in its high-spin +II oxidation state [
26,
27]; this is particularly so in pseudotetrahedral symmetry. Several parameters can potentially be employed to stabilize easy-axis (or Ising-type) magnetic anisotropy including the softness (HSAB) of the donor atoms (and hence the Co
II–ligand covalency), the influence of the other commonly used coligands (e.g., halides) and the variation of the counter cation in anionic complexes. Representative examples are briefly described below.
The first transition-metal SIM without requiring the application of an external dc field to suppress quantum relaxation processes was the tetrahedral complex (Ph
4P)
2[Co(SPh)
4] (
17) [
104], prepared as illustrated in Equation (12). The complex has an
S = 3/2 spin ground state, with a large, negative, axial zero-field splitting (
D = −70 cm
−1) and a low rhombicity (
E/
D < 0.9). The large magnetic anisotropy of the anion can be explained by examining
Figure 13, derived from angular overlap model calculations. The near degeneracy of the filled 3d
x2−y2 orbital and the singly occupied 3d
xy orbital leads to a low-lying electronic excited state that can couple to the ground electronic state through spin-orbit coupling, thus affording a large
D value and resulting in a
Ueff value of 21(1) cm
−1. Dilution of [Co(SPh)
4]
2− within the isostructural [Zn(SPh)
4]
2− matrix eliminates quantum tunneling pathways, indicating that they occur via intermolecular dipolar interactions [
104].
Continuing the above mentioned work, the group of Long prepared the salts of [Co(EPh)
4]
2− (E = O, Se) (Ph
4P)[Co(OPh)
4] (
18), K(Ph
4P)[Co(OPh)
4] (
18a), (Ph
4P)
2[Co(SePh)
4] (
19), from reactions of CoCl
2 or CoI
2 with excess amounts of K(EPh) and (Ph
4P)Br in MeCN [
105]. All anions possess pseudotetrahedral coordination environments with tetragonal distortions to give exactly or approximately D
2d symmetry. One of the goals was to correlate
D and
Ueff. The values of the former are ~−11, −24 and −83 cm
−1 for
18,
18a and
19, respectively. Arrhenius plots of the ac data indicate
Ueff values of 21(1) and 19(1) cm
−1 for
18 and
19, respectively, whereas the plot for
18a shows substantial curvature indicating strong intermolecular interactions. Dilution experiments with [Zn(OPh)
4]
2− allowed the observation of thermally-activated magnetic relaxation for
18a with a
Ueff value of 34.0(5) cm
−1. The trend in
Ueff for
17,
18,
18a and
19 does not follow the trend in
D values; this possibly indicates that magnetization relaxation in
17,
18a and
19 is not fully thermally activated (the relation between
D and
U for an
S = 3/2 system is
U = 2
D). An analysis of the four complexes within the framework of ligand field theory shows that the increase in |
D| takes place in concert with a decrease in the Racah parameter
B, i.e., increased covalency. This suggest the importance of soft donor ligands in the efforts to obtain systems with a large magnetic anisotropy.
The story does not end here. Inspired from the above described studies, the groups of Neese and Atanasov reported a systematic theoretical study developing magnetostructural correlations in the anions [Co
II(EPh)
4]
2− (E = O, S, Se, Te; the Te derivative is hypothetical) based on multireference quantum chemical methods and ab initio-based ligand field theory; they also discussed the correlation of
D with softness of the ligands, relativistic nephelauxetic effects and covalency [
106]. The |
D| value increases when the ligand field decreases across the series from O to Te. It has been shown that due to the π-anisotropy of the S and Se donor atoms, magnetostructural correlations in [Co(OPh)
4]
2− and [Co(EPh)
4]
2− (E = S, Se) differ. In the case of the isotropic PhO
− ligand, only variations within the first coordination sphere of Co
II affect magnetic properties; in the case of the PhE
− (E = S, Se) ligands, variations in the first and second coordination sphere affect equally the magnetic properties. The influence of the counter cations on the spin Hamitonian parameters was also studied in the two salts (Ph
4P)
2[Co(SPh)
4] (
17; D
2d symmetry) and (Et
4N)
2[Co(SPh)
4] (
17a; S
4 symmetry). The characterization techniques employed were high-field/high-frequency EPR, multifield SQUID magnetometry, frequency domain Fourier-transform THz-EPR and variable-field variable-temperature magnetic circular dichroism [
107]. The [Co(SPh)
4]
2− anion in
17 shows strong axial magnetic anisotropy as already was known [
104,
105], whereas the anion in
17a shows rhombic anisotropy with
D = +11 cm
−1 and
E/
D = ~0.20 [
107]. It has been verified, also with the help of multireference ab initio calculations, that the differences observed in the two complexes are associated with slight changes of the S–Co–S bond angles and C–S–Co–S torsion angles around the {Co
IIS
4} unit.
Another excellent experimental study on mononuclear Co(II) complexes is in agreement with the fact that ligands with heavier and softer main group donor atoms increase the magnetic anisotropy of the complexes, as evidenced by the increased |
D| values [
108]. The reactions of CoI
2 and the monodentate ligands quinoline (qun) and Ph
3P in anhydrous EtOH, and Ph
3As in MeNO
2, all under refluxing conditions, give complexes [CoI
2(qun)
2] (
20), [CoI
2(Ph
3P)
2] (
21) and [CoI
2(Ph
3As)
2] (
22) in good yields. The crystal structures of the complexes reveal a pseudo-tetrahedral local coordination environment around the central Co
II atom. The
D value was found to vary from +9.2 cm
−1 in
20 to −37 cm
−1 in
21 and −75 cm
−1 in
22. However, the dynamic properties reveal only a minor effect on the
Ueff value. Compound
20 does not show an out-of-phase ac magnetic susceptibility signal under a zero or applied dc field; complexes
21 and
22 exhibit slow magnetization relaxation below 4 K under an applied dc field of 1000 Oe with
Ueff values of ~21 and ~23 cm
−1, respectively. It is obvious that the observed increase in the energy barrier for the As-based complex
22 (~−21 cm
−1→~−23 cm
−1) is much smaller than the corresponding increase in the D value (~−37 cm
−1→~−75 cm
−1).
Analogous studies have been performed by the groups of Rajaraman and Shanmugam [
109]. They used the exocyclic mesoionic ligands 2,3-diphenyl-1,2,3,4-tetrazolium-5-olate (L
1) and 2,3-diphenyl-1,2,3,4-tetrazolium-5-thiolate (L
2), whose general structural formula is shown in
Figure 14. Mesoionic ligands contain dipolar 5- or 6-membered rings whose canonical resonance structures cannot be represented without any additional charges in them. Their coordination chemistry is almost completely unexplored. Such ligands offer flexibility which allows researchers to selectively change the coordinating substituents and, thus, to investigate the influence of the donor atoms on the magnetic anisotropy. The 1:1 reactions of CoX
2∙2H
2O (X = Cl, Br) with L
1 or L
2 in MeOH provide access to blue complexes [CoX
2(L
1)(MeCN)] (X = Cl,
23a; X = Br,
23b) or green [CoX
2(L
2)(MeCN)] (X = Cl,
24a; X = Br,
24b) when crystallized from MeCN; the preparation of
23a and
23b is illustrated in Equation (13). The complexes are pseudotetrahedral, the donor atoms of monodentate L
1 and L
2 being the exocyclic oxygen and sulfur atoms, respectively. The
D values, deduced from magnetization data, are +15.6 cm
−1 (
23a), +11.2 cm
−1 (
23b), −11.3 cm
−1 (
24a) and −10.3 cm
−1 (
24b). Thus, simple substitution of L
1 (O-donor in
23a and
23b) by L
2 (S-donor in
24a and
24b) switches the single-ion magnetic anisotropy parameter from positive to negative. All four complexes are field-induced SIMs with
Ueff values of 10.3 cm
−1 (
23a), 8.2 cm
−1 (
23b), 20.2 cm
−1 (
24a) and 13.8 cm
−1 (
24b).
8. Combined Metallacrown and Click Chemistry as a Tool for SMM Research
Metallacrowns (MCs) are analogues of crown ethers. They consist of a repeat unit of –{M’–N–O}
n– in a cyclic arrangement where the ring metal ion and the nitrogen atom replace the methylene carbon atoms of a crown ether. MCs are named and abbreviated on the basis of the ring size and the number of oxygen atoms that act as donors. For example, in the abbreviation 12-MC-4, MC represents a metallacrown that is a 12-membered ring comprising 4 repeating –{M’–N–O}– units with 4 donating oxygen atoms. The nomenclature/abbreviation also includes the bound central metal ion M, the ligand, and any bound or unbound ions. Thus, in the typical representation [MX{ring size-MC
M’Z(L)-ring oxygens}]Y, M is the bound central metal with its oxidation state, X is any bound anion, M’ is the ring metal with its oxidation state, Z is the third heteroatom of the ring (usually N), L is the organic ligand of the complex, and Y is any unbound anion. Sometimes there are unbound cations, which are placed before the bound central metal M. An example of the above naming scheme is [Gd
III(NO
3)
2{15-MC
CuIIN(picha)-5}](NO
3), where H
2picha is picoline hydroxamic acid. The molecular structure of the hexanuclear cation is shown in
Figure 15. The –{M’–N–O}– repeat unit is now a quite general motif in inorganic chemistry and the connectivity has significantly grown to include a variety of bridges such as –{N–N}–, –{O–P}–, –{N–C–O}–, –{N–C–N}–, –{O–C–O}– and –{X}– (X is a nonmetal). After the initial great efforts to synthesize many MCs and to discover a great variety of structural types, the research activity in the past 15 years or so has shifted towards the use of these unique compounds for applications, e.g., for selective binding of cations or anions, as MRI contrast agents, in catalysis, as mimics of surface science, as building blocks for 1-, 2- and 3-dimensional solids, in liquid crystals and in various aspects of Molecular Magnetism [
110]. Metallacrown chemistry has a brilliant potential for growth.
In addition to its use in inorganic chemistry (
Section 4) as an anion, the azide functional group was widely used after the second world war in the synthesis of N-containing natural products and medicinal formulae. For example, the azide group is one of the most efficient amine precursors, and its ability to undergo 1,3-dipolar cycloadditions and diazo-transfer reactions is a valuable tool in organic synthesis. The [3 + 2] cycloadditions between azides and alkynes were first observed by Michael in 1893 and examined later by Huisgen [
111]. The reaction has a high activation energy barrier and elevated temperatures or pressures are required to facilitate it. Almost 20 years ago, it was discovered that Cu(I) catalysis accelerates the rate of formal cycloaddition between azides and terminal alkynes, which proceeds at ambient temperatures and pressures affording 1,4-disubstitued 1,2,3-triazoles exclusively. This variant of the Huisgen cycloaddition has been termed CuAAC for
Cu-catalyzed
azide
alkyne
cycloaddition. CuAAC is an exciting example of “click chemistry”, a term used by Sharpless and coworkers to describe a category of chemical reactions that link two components in high yields and with minimal byproducts [
112].
The group of Rentschler has developed an approach which allows for the rational decoration of Cu(II) MCs with SIMs or SMMs using click chemistry [
113]. The CuAAC is suitable for covalently linking magnetic building blocks which bear azide and terminal alkyne functional groups. Under mild conditions, the process leads selectively to a 1,4-disubstituted 1,2,3-triazole as a conjugated bridge which ensures communication between two or more magnetic building blocks. In order for the approach to be successful, several conditions must be satisfied: (i) To isolate thermodynamically stable complexes, polydentate chelating ligands are required. (ii) The number of introduced modified ligands must be limited to prevent an overloading of the complex. (iii) A short distance between the metal ions and the periphery of the ligands, as well as a conjugated π system are important to enable strong magnetic communication between the building blocks; and (iv) The latter should preserve their structural integrity in the reaction solvent. It is worth mentioning that this click concept can be further used in other challenging research areas of Molecular Magnetism, e.g., covalent anchoring of SMMs on surfaces, development of new cluster-based inorganic-organic hybrid materials, etc. We give an example of this strategy.
The two functionalized complexes are (Me
4N)
2[Cu
II{12-MC
Cu(II)N(eshi)-4}] (
25) [
114] and [Co
II(oda)(aterpy)] (
26) [
115]. Compound
25 was prepared by the high-yield reaction illustrated in Equation (14), where H
3eshi is 4-ethynylsalicylhydroxamic acid. In the centrosymmetric pentanuclear dianion (
Figure 16), each of the Cu
II atoms has an almost square planar coordination geometry. The peripheral four metal ions are bridged by N–O groups of the eshi
3− ligands forming a 12-membered metallacrown planar ring, which encapsulates the central Cu
II site through coordination of the μ
2 oxygen atoms of the N–O groups. The other two, terminally ligated oxygen atoms of each eshi
3− ligand complete coordination at neighboring Cu
II atoms. The four alkynyl groups protrude from the planar cluster dianion and each of them is orthogonal to its two neighbors. The complex exhibits an isolated
S = 1/2 ground state with
J1 = −158 cm
−1,
J2 = −65 cm
−1 and
g = 2.13, adopting the Heisenberg Hamiltonian Σ
ij (−2
Jij∙
Ŝi∙
Ŝj);
J1 and
J2 are the coupling constants corresponding to the radial magnetic interaction between the central Cu
II atom and the ring Cu
II atoms, and the tangential exchange interaction between two neighboring ring metal ions, respectively. The solution integrity of the pentanuclear dianion of
25 is proven by paramagnetic 1D and 2D
1H NMR spectroscopy in d
6-DMSO and ESI-MS studies in a DMSO/MeOH solvent matrix. These results suggest that the 12-MC-4 Cu(II) complex is highly stable and thus suitable to perform CuAAC click reactions.
Compound
26 was prepared [
115] by the reaction illustrated in Equation (15) in ~30% yield; the neutral ligand aterpy is 4′-azido-2,2′:6′,2′’-terpyridine and oda is the oxodiacetate(−2) ligand. Complex [Zn(oda)(terpy)] (
26a) was prepared in a similar manner (yield ~50%). The two complexes are isomorphous. The metal ion is in an octahedral coordination environment with rhombic (C
2) distortion because of the rigidity of the planar, tridentate chelating, tripodal aterpy and oda
2− ligands (
Figure 16). The chelate effect of the two ligands provides the complexes with high thermodynamic stability in DMSO, as proven by ESI-MS and
1H NMR (for the diamagnetic complex
26a) studies. Thus, both complexes are suitable for CuAAC click reactions. Ac susceptibility data under a static dc field of 0.15 T reveal that the mononuclear Co(II) complex is a weak SIM.
The click reactions of
25 with
26 or
26a in d
6-DMSO at 80 °C with copper(I) iodide as catalyst lead to complexes (Me
4N)
2[Cu
II{12-MC
Cu(II)N([M(II)(oda)(ttshi)]-4}] (
27, M = Co;
27a, M = Zn) [
113,
114], Equation (16) and
Figure 15; H
3ttshi is 4-(2,2′:6′,2′’-terpyridyl-1H-[
1,
2,
3]-triazol-4-yl)salicylhydroxamic acid. The nature and structural type of the complexes were verified by IR and UV/VIS spectroscopic techniques, ESI-MS studies, as well as by paramagnetic
1H NMR methods for the {Cu
5Zn
4} cluster
27a. Magnetic studies of
27a indicate fairly strong antiferromagnetic Cu
II∙∙∙Cu
II exchange interactions and an
S = 1/2 ground state, similar to the behavior of the precursor complex
25. Orbital contributions and spin-orbit coupling effects from the additional Co
II atoms make difficult the analysis of the magnetic properties of
27. The important fact, however, is that ac susceptibility measurements of this complex reveal a weak out-of-phase signal at low temperatures and low-field oscillation frequencies. This is the first time (and a very promising approach) where the CuAAC click chemistry has been used to upgrade the magnetic properties of a simple copper(II) metallacrown with remarkable magnetic features from the attached Co(II) SIM units, opening new doors for the creation of interesting molecular magnetic materials and providing a contribution toward the future development of SMM-based quantum computers [
113]; the latter require linking of SMMs/SIMs in a deliberate manner, like the click chemistry approach presented here. Whatever the future form of quantum technology will be, it is likely that the role of chemists will be the design and optimization of molecules that couple to an external stimulus in the adequate energy rate, while offering some elementar functionality [
37]. SMMs/SIMs, being much more versatile than magnetic atoms, and yet microscopic are among the quantum objects with the highest capacity to form non-trivial ordered states at the nanoscale and to be replicated in large numbers by means of chemical methods.
9. Deprotonated 2-Pyridyl Alcohols: Central “Players” in the Chemistry of Transition-Metal SMMs
When developing routes and strategies for the synthesis of transition-metal SMMs, the design of bridging ligands is of paramount importance. In addition to bridging ability, the propagation of strong magnetic exchange interaction between the metal spin carriers and the simultaneous formation of chelating rings are highly desirable. The anions of 2-pyridyl alcohols (
Figure 17) have proven to be versatile chelating and bridging groups that have yielded a number of 3d-metal, especially Mn, and 3d/4f-metal clusters with various structural motifs [
116,
117,
118,
119,
120,
121,
122,
123,
124,
125,
126,
127,
128,
129,
130,
131,
132,
133]. Moreover, the bridging deprotonated oxygen atom, i.e., the alkoxido group, often supports ferromagnetic coupling between the metal ions and has thus yielded polynuclear complexes with large
S values and SMM properties. We give below examples from the use of such ligands in 3d-metal SMM chemistry.
The 1:1:1 NiCl
2∙6H
2O/Hhmp/NaOR reaction mixtures in alcohols (MeOH, EtOH) give clusters [Ni
4(hmp)
4Cl
4(ROH)
4] (
28, R = Me;
28a, R = Et), Equation (17); hmp is the anion of 2-(hydroxymethyl)pyridine (R = R’ = H and
n = 1 in
Figure 17). The tetranuclear cluster molecules possess a distorted cubane {Ni
4(μ
3-OR’’)
4}
4+ core, where R″– = (2-pyridyl)CH
2–, with the Ni
II atoms and the oxygen atoms from the 3.31 hmp
− ligands (
B with R = R’ = H and
n = 1 in
Figure 4) occupying alternate vertices of the cube. A terminal chlorido ligand, an alcohol molecule and a 2-pyridyl nitrogen atom complete the octahedral coordination sphere of each metal ion [
121]. Variable-temperature dc magnetic susceptibility studies indicate ferromagnetic Ni
II∙∙∙Ni
II exchange interactions and a
S = 4 ground state. Single-crystal high-frequency EPR spectra clearly suggest that each of the complexes has a total spin of 4 in the ground state with a negative
D value. Magnetization vs. magnetic field measurements performed on single crystals with a micro-SQUID magnetometer show that these {Ni
4} clusters are SMMs. An appreciable exchange bias is evident in the hysteresis loops. The first resonant tunneling step is shifted considerably from zero field by virtue of intermolecular antiferromagnetic exchange interactions between the tetranuclear molecules of
28 and
28a in their crystals [
121].
Deprotonated 2-pyridyl alcohols have contributed a lot into the development of Mn SMMs. For example, the reaction of Hhmp with Mn(O
2CPh)
2 in the presence of Et
3N in CH
2Cl
2/MeOH leads to the isolation of [Mn
II6Mn
III10O
8(OH)
2(O
2CPh)
12(hmp)
10(H
2O)
2](O
2CPh)
2 (
29) in 30% yield, Equation (18). The core structure consists of a linked pair of complete cubanes {Mn
II2Mn
III2(μ
4-O)
2(μ
3-OR’’)
2}
4+, on either side of which is attached a tetrahedral {Mn
IIMn
III3(μ
4-O)}
9+ unit. Among the ten hmp
− groups, four are bridging within the central cubanes in a 3.31 mode (
B with R = R’ = H and
n = 1 in
Figure 4) and the other six are bridging within the outer tetrahedral units in a 2.21 mod (
C with R = R’ = H and
n = 1 in
Figure 4). The Mn ions are all six-coordinate with distorted octahedral geometry, except for the Mn
II atoms of the tetrahedral units which are 7-coordinate. Solid-state dc and ac magnetic susceptibility measurements on
29 establish that it possesses a
S = 8 ground state. The complex displays frequency-dependent out-of-phase (
χ″
M) ac susceptibility signals below 3 K suggestive of SMM behavior. The
D and
Ueff value are −0.11 cm
−1 and 7 cm
−1, respectively. Magnetization vs. applied dc field sweeps on single crystals of the complex down to 0.04 K exhibit hysteresis, confirming
29 to be a SMM, albeit weak [
131]. Comparison of the structure of
29 ({Mn
16}) with {Mn
12} and {Mn
6} clusters obtained under the same reaction conditions but with two Me (R = R’ = Me and
n = 1 in
Figure 17) or two Ph (R = R’ = Ph and
n = 1 in
Figure 17) groups, respectively, added next to the alkoxido O atom of hmp
− indicate their influence on the nuclearity and structure of the products [
130,
131] as being due to the overall bulk of the ligand plus the decreased ability of the deprotonated oxygen atom to bridge.
The ligand Hhep (R = R’ = H and
n = 2 in
Figure 17) also gives structurally and magnetically interesting clusters. The reaction of a 2:1 mixture of [Mn
III3O(O
2CMe)
6(py)
3](ClO
4) and [Mn
II,III,III3O(O
2CMe)
6(py)
3] with 4.5 equivs. of Hhep in MeCN affords cluster [Mn
II2Mn
III16O
14(O
2CMe)
18(hep)
4(Hhep)
2(H
2O)
2](ClO
4)
2 (
30) in 20% yield. The core appears to be {Mn
18(μ
4-O)
4(μ
3-O)
10(μ
3-O
acetato)
2(μ
2-acetato)
2(μ
2-O
hep¯/Hhep)
6}
14+ [
119,
125]. It can be described as a central {Mn
4O
6} unit (containing a linear Mn
4 chain) linked by its μ
3-O
2− ions to two {Mn
7O
9} units, one on each side. Each of the heptanuclear units comprises a face-sharing set of one {Mn
4O
4} cubane and two {Mn
3O
4} partial cubanes. All the metal ions are six-coordinate. The hep
− and Hhep groups behave as 2.21 (
C with R = R’ = H and
n = 2 for hep
− and
D for Hhep in
Figure 4) ligands. Fitting of magnetization data establish that
30 possesses a total spin of 13 in the ground state and a
D value of -13 cm
−1. The complex is SMM (
Ueff = 15 cm
−1), and this is confirmed by the appearance of hysteresis loops in magnetization vs. dc field sweeps on a single crystal. Below 0.2 K, the relaxation becomes temperature-independent, consistent with relaxation only by QTM through the anisotropy barrier via the lowest-energy
MS = ±13 levels of the
S = 13 spin manifold. Although the high nuclearity of the complex is mostly due to the presence of 4.4- and 3.3-O
2− groups, as well as 4.31, 2.21 and 2.11 MeCO
2− ligands, the 2.21 hep
− and Hhep moieties certainly contribute into its interesting magnetic properties.
2-pyridyl alcohols can be chiral leading to 3d-metal clusters with unprecedented structural motifs and interesting magnetic properties. The ligand α-methyl-2-pyridine-methanol (Hmpm; R = H, R’ = Me and
n = 1 in
Figure 17) presents similar coordination features to Hhmp, but offers slightly different steric and electronic effects. The Hmpm ligand can be prepared via the reduction of 2-acetylpyridine by NaBH
4 [
133]. The 2:1:2 reaction between Mn(O
2CPh)
2∙2H
2O, rac-Hmpm and Et
3N in MeOH gives a deep red solution, which upon slow solvent evaporation at room temperature affords cluster [Mn
II2Mn
III28Mn
IV(OH)
2(OMe)
24O
24(O
2CPh)
16(rac-mpm)
2] (
31) in ~30% yield, Equation (19). The core (
Figure 18, left) can be described as a consecutive array of edge-sharing {Mn
4(μ
4-O)} tetrahedra and {Mn
3(μ
3-O)} triangles that are linked to each other via bridging MeO
− and O
2− groups [
133]. An alternative description of the core is as consisting of seven parallel layers (
Figure 18, right) of four types (A, B, C, D) with an ABCDCBA arrangement. Layers A and B are simple Mn
III monomeric and {Mn
III4} butterfly subunits, respectively, attached to each other through a 4.4 oxido group. Layer C is a {Mn
IIMn
III3} cluster moiety containing three edge-sharing {Mn
3} triangles. Layer D comprises a {Mn
III8Mn
IV} rod-like cluster unit that can be further seen as a central, planar disk-like {Mn
III6Mn
IV} moiety with two additional Mn
III atoms above and below the disk. The layers are held together by a combination of bridging methoxido and oxido groups. The rac-mpm
− groups behave as 2.21 ligands (
C with R = H, R’ = Me and
n = 1 in
Figure 4). The molecule is spherical with a diameter of ~2.5 nm. The cluster is: (i) One of the largest 3d-metal clusters, (ii) the second highest-nuclearity Mn complex containing an odd number of metal centers and the second cluster with a nuclearity of 31, and (iii) the largest SMM (
S = 23/2,
Ueff = ~40 cm
−1) that possesses entirely resolved out-of-phase ac peaks and magnetization hysteresis loops below 5 K.
10. Deprotonated Aliphatic Diols: Simple and Efficient Ligands in the Chemistry of 3d-Metal SMMs
Molecules with two hydroxyl groups on different aliphatic carbon atoms have some interesting chemical characteristics [
134,
135] which are more prominent for vicinal diols (
vic-diols). In the last 15 years or so, there has been an intense interest in the use of diols in the chemistry of transition-metal clusters and SMMs [
136]. The simplistic rationale here is that each deprotonated alkoxido oxygen atom can bridge two or three metal ions and the latter can be linked together into polymetallic arrays with hopefully large ground-state spins, anisotropies and SMM properties. The structures of the products depend on (i) the level of deprotonation of the ligand (singly or doubly deprotonated), (ii) the existence of other donor groups on the diol (e.g., a pyridyl group, –NH
2, –COOH, …), and (iii) the presence of other bridging co-ligands in the reaction system. A family of simple aliphatic diols, with no other donor groups, consist of 1,3-propanediol and its derivatives (
Figure 19). We give below one notable example from the use of a member of this family in Mn SMM chemistry. The cited complexes belong to the class of giant homometallic 3d clusters (nuclearities higher than 30) with the metals in moderate oxidation states. Such giant clusters are of great interest not only for their impressive structures with nano dimensions, but also because they often display interesting magnetic properties (including SMM behavior). In addition, they possess the properties of both classical and quantum world, thus giving the opportunity for the discovery of new physical phenomena and the deeper understanding of the existing ones [
137].
The 1:4:1 reaction of [Mn
II,III,III3O(O
2CMe)
6(py)
3] with H
2pd and NaN
3 in MeCN leads to cluster [{Mn
II2Mn
III8NaO
2(O
2CMe)
13(pd)
6(py)
2}
4] (
32) in 35% yield [
138,
139], Equation (20). Since the giant cluster contains Na
+ but not N
3−, it has been assumed that NaN
3 is important as the source of Na
+ and perhaps to provide additional weak base (N
3−) for ligand deprotonation and further oxide ion formation; however, the main sources for ligand deprotonation are the MeCO
2− ions, as shown in Equation (20), and/or the pyridine molecules. This is supported by the same reactions, but using NaCN, NaOCN, NaSCN or Na{N(CN)
2} instead of NaN
3, which all give
32 in slightly lower yields (18–26%). The molecule of
32 consists of four {Mn
II2Mn
III8} loops linked through four Na
+ ions to give a supramolecular aggregate with a saddle-like topology; one loop with one Na
+ ion is shown in
Figure 20. The Mn ions are all in a distorted octahedral coordination environment; the eight high-spin 3d
4 Mn
III centers display the Jahn–Teller axial elongation, but the Jahn–Teller axes are not co-parallel. Each loop consists of two triangular {Mn
III3(μ
3-O)}
7+ and two dinuclear {Mn
IIMn
III} subunits linked by the oxygen atoms of the six 3.22 pd
2− groups (
E with R = R’ = H in
Figure 4), as well as by 2.11 and 4.22 MeCO
2− ligands. The triangular subunits are connected by two pd
2− oxygen atoms, whereas the dinuclear subunits are connected by two pd
2− oxygen atoms and a 2.11 MeCO
2− ligand. The Mn
II and Mn
III atoms within each dinuclear subunit are bridged by a 2.11 acetato group, a pd
2− oxygen atom and an oxygen atom from a 4.22 MeCO
2− ligand. The Mn
III atoms of each triangular subunit are bridged by a 3.3 O
2− group, two oxygen atoms from two different pd
2− groups, one oxygen atom from a 4.22 MeCO
2− ligand and two 3.21 MeCO
2− ligands. The latter and an additional acetato group link each triangular subunit to a Na
+ ion; the two alkali metal ions attached to the decanuclear {Mn
II2Mn
III8} loop connect it in an equivalent manner to a neighboring loop giving a giant loop-of-loops aggregate. Four of the pd
2− groups bridge one Mn
II and two Mn
III centers, and the remaining two bridge three Mn
III ions. The crystal structure shows that the {Mn
II8Mn
III32Na
4} aggregates pack as tail-to-tail {Mn
II8Mn
III32Na
4}
2 dimers, thus leading to egg-shaped stacks. A detailed magnetic study of
32 reveals that each decanuclear loop has a
S = 4 ground-state spin, and displays frequency-dependent in-phase and out-of-phase ac magnetic susceptibility signals. The aggregate also exhibits hysteresis loops. The hysteresis loops are not typical of SMM behavior because of the presence of interloop exchange interactions through the diamagnetic Na
+ ions, and also intermolecular interactions between different {Mn
II8Mn
III32Na
4} aggregates [
137,
139].
Once the heterometallic (Na/Mn) character of
32 and its influence on the magnetic properties had been studied, the synthesis of the magnetically discrete, homometallic {Mn
44} analogue of
32 was sought, as a means of strengthening the interloop exchange interactions and appearance of better SMM properties. The desired product [Mn
II12Mn
III32O
8(O
2CMe)
52(pd)
24(py)
8]
4+ was isolated [
139] from the 1:5:1 [Mn
II,III,III3O(py)
3]/H
2pd/Mn
II(ClO
4)
2∙6H
2O reaction mixture in CH
2Cl
2, Equation (21), i.e., with the use of Mn
II(ClO
4)
2∙6H
2O instead of NaN
3. The product is, as expected, cationic with the formula [Mn
II12Mn
III32O
8(O
2CMe)
52(pd)
24(py)
8](OH)(ClO
4)
3 (
33) and its yield is ~25%. The molecular structure of the cation of
33 is very similar to that of the molecule of
32, the main difference being the fact that the {Mn
II2Mn
III8} loops in the former are linked through Mn
II ions, whereas those of the latter by Na
+ ions; as a result the {Mn
44} cluster is positively charged, whereas the {Mn
40Na
4} aggregate is neutral. The packing of
33 is similar to that in
32, and thus supramolecular {Mn
44}
2 dimers are formed in the crystal. In accord with the strongest magnetic exchange interactions between the four {Mn
II2Mn
III8} loops mediated through the connecting Mn
II centers, magnetic susceptibility studies reveal that
33 has a
S = 6 ground-state spin and displays frequency-dependent in-phase and out-of-phase signals. Magnetization vs. dc magnetic field sweeps on single crystals of
33 display hysteresis loops below 0.7 K whose coercivities increase with decreasing temperature and with increasing sweep rate, confirming that this giant cluster is one of the largest Mn SMMs.
The ligand 2-methyl-1,3-propanediol (H
2mpd; R = H and R’ = Me in
Figure 19) gives the heterometallic aggregate [{Mn
II2Mn
III8NaO
2(O
2CMe)
13(mpd)
6(py)
2}
4] (
34) [
139], which is structurally and magnetically similar to
32. The ligand pd
2− has been also used by Tasiopoulos’ group to construct giant Mn/Co
II and Mn/Ni
II clusters with exciting structures and large ground-state spins, but with no SMM properties [
140,
141]. For example, cluster [Mn
II8Mn
III28Ni
II4O
12Cl
10(O
2CMe)
26(pd)
24(py)
4(H
2O)
2] (
35) possesses an unprecedented “loop-of loops-and-supertetrahedra” structural topology and displays a high ground-state spin state value of 26 ± 1 [
141].
11. Molecular and Supramolecular Approaches in the Chemistry of Manganese SMMs Using Simple and Elaborate 2-Pyridyl Oximes
The deprotonated oxime (oximate, R
2CNO
−) group has played an important role in the chemistry of 3d-metal clusters and SMMs. This diatomic group can bridge up to three metal ions and often propagates ferromagnetic exchange interactions; the latter property may lead to SMM properties. In most cases, the oxime group is part of an organic ligand that possesses one or more other donor groups, often in a position that enables the formation of a stable chelating ring with the participation of the oximate nitrogen atom. The two most studied families of oxime groups in Molecular Magnetism are the salicyl aldo(keto)ximes and the 2-pyridyl aldo(keto)ximes,
Figure 21. The former [
142,
143,
144,
145,
146,
147,
148,
149,
150,
151,
152,
153,
154,
155] have led to a variety of clusters with exciting molecular structures and magnetic properties; they have been used, among others, in the development of a synthetic process widely known as “ground-state spin switching and enhancing SMM properties via targeted structural distortion” strategy [
56,
142,
143,
144,
145,
147,
148,
149]. The latter [
156,
157,
158,
159,
160,
161,
162,
163,
164,
165,
166,
167,
168,
169,
170,
171,
172,
173,
174], in addition to their involvement in the synthesis of 3d-metal SIMs [
158], have provided access to interesting 3d-metal clusters and SMMs. They have also been used in the realization of a synthetic scheme best known as “‘switching on’ SMM properties upon conversion of low-spin complexes into high-spin ones without changing the core” strategy [
168,
169] and, when derivatized by design, in the development of an innovative approach which allows the covalent linking of SMMs by applying principles of supramolecular chemistry [
172,
173,
174]. Below we describe briefly the molecular and supramolecular approaches in the chemistry of Mn SMMs by using 2-pyridyl ketoximes and its derivatives as key synthetic tools.
An in-depth studied family of Mn(III) carboxylate clusters consists of the triply oxido-bridged, triangular complexes [Mn
3O(O
2CR’’)
6L
3]X (R’’ = Me, Et, Ph, …; X = monoanionic counterions; L = neutral monodentate ligands) [
175]. These complexes possess the {Mn
III3(μ
3-O)}
7+ core with peripheral ligation provided by 2.11 carboxylato groups and terminal L ligands. The triangular cations are characterized by antiferromagnetic Mn
III∙∙∙Mn
III exchange interactions and have low
S values in the ground state; they are not thus SMMs. It was a general belief that this common triangular topology could never give complexes with SMM properties. Using 2-pyridyl ketoximes, however, these complexes can be converted into triangular clusters with the same {Mn
3(μ
3-O)}
7+ core, but with ferromagnetic Mn
III∙∙∙Mn
III interactions. The strategy is illustrated in
Figure 22. The 1:3 reaction between [Mn
3O(O
2CR’’)
6(py)
3](ClO
4) (R’’ = Me,
36a; R’’ = Et,
36b; R’’ = Ph,
36c, see
Figure 22, left) and methyl 2-pyridyl ketoxime (Hmpko; R = Me and R’ = H in
Figure 21) in MeCN/MeOH give dark brown solutions; evaporation of the reaction solutions to dryness and crystallization of the residues from CH
2Cl
2/n-hexane give dark red crystals of [Mn
3O(O
2CR’’)
3(mpko)
3](ClO
4) (R’’ = Me,
37a; R’’ = Et,
37b; R’’ = Ph,
37c) in high yields (>80%), Equation (22) [
168]. The 1:3 molar ratio of the reactants was chosen to allow for the incorporation of one mpko
− ligand onto each edge of the {Mn
3(μ
3-O)}
7+ core. The reaction can thus be described as a simple ligand substitution with the replacement of three R’’CO
2− groups and three py ligands by three mpko
− ones, without change of the Mn oxidation level. This reaction scheme is quite general and can be extended to other carboxylate groups and 2-pyridyl ketoximes (
Figure 21), where R is a non-donor group.
As in
36a,
36b,
36c, the cations of
37a,
37b,
37c (
Figure 22, right) possess a {Mn
III3(μ
3-O)}
7+ triangular core, but each Mn
2 edge is now bridged by a 2.11 R’’CO
2− and a 2.111 mpko
− (
G with R = Me and R’ = H in
Figure 4) group. The three R’’CO
2− groups lie on one side of the plane defined by the Mn
III atoms and the three oximato groups on the other. The tridentate ligation mode of mpko
− causes a buckling of the formerly planar {Mn
III3(μ
3-O)}
7+ unit, giving rise to a relative twisting of the octahedra of the metal ions, a slight non-planarity of the Mn
III–N–O–Mn
III units and a displacement of the central oxido group which is ~0.3 Å above the {Mn
III3} plane on the same side as the carboxylate groups. These structural distortions lead to ferromagnetic Mn
III∙∙∙Mn
III interactions resulting in a
S = 6 ground state. Fits of dc magnetization data collected in the 10–70 KG and 1.8–10.0 K ranges confirm the ground-state spin and give a
D value of ~−0.35 cm
−1. Complexes
37 exhibit frequency-dependent out-of-phase (χ″
M) ac magnetic susceptibility signals suggesting a possible SMM behavior. Relaxation rate vs.
T data down to 1.8 K obtained from the χ″
Μ vs.
T studies were supplemented with rate vs.
T data measured to 0.04 K via magnetization vs. time decay studies, and these were used to construct Arrhenius plots from which
Ueff values of ~8 cm
−1 were derived. Magnetization vs. dc field sweeps on single crystals of
37a∙3CH
2Cl
2 show hysteresis loops which exhibit steps due to QTM. The loops are temperature-independent below 0.3 K and this indicates only ground-state QTM between the
MS = ±6 levels. Complexes
37 were the first confirmed triangular SMMs of any transition metal. High-frequency EPR spectra of single crystals of
37a∙3CH
2Cl
2 give
D = −0.3 cm
−1 and provide evidence of a rather significant transverse anisotropy (|
E| ≥ 0.015 cm
−1). DFT calculations provide strong evidence [
169] that the unusual ferromagnetic exchange interactions in complexes
37 originate from a combination of several factors including the non-planarity (with respect to the {Mn
III3} plane) of the bridging oximato groups, the non-parallel alignment of the Jahn–Teller axes and the shift of the triply-bridging oxido group out of the plane defined by the three Mn
III ions. The above results demonstrate that structural distortions of a magnetic core imposed by peripheral ligands (deprotonated 2-pyridyl oximes in this case) can “switch on” SMM properties [
168].
Ligands containing two 2-pyridyl ketoxime moieties are of special interest in the realm of the linking of SMMs [
172,
173,
174]. Since SMMs have been proposed as qubits for quantum information processes and as components in molecular spintronics, a great challenge is their quantum mechanical coupling to each other or to other components of a device, while maintaining the intrinsic properties of each SMM; this maintenance requires very weak coupling between the SMM precursors. Excluding utilization of hydrogen bonds, with which it is difficult to control oligomerization and to achieve retention of the supramolecular structure in solution, the best solution is the designed linking of SMMs via coordination bonds. The groups of Papaefstathiou, Escuer, Brechin and Christou [
150,
151,
152,
153,
170,
171,
172,
173,
174], among others, have used building-block strategies to link SMMs together employing carefully chosen linkers that provide inter-SMM interactions and ensure the formation of discrete oligomeric (and not polymeric) species.
The SMM cations of
37a,
37b and
37c (
Figure 22, right) are excellent candidates to be used as building blocks for such a strategy. They have their R’’CO
2− and mpko
− ligands on opposite sites of the {Mn
III3} plane. The tripodal arrangement of the carboxylato and oximato groups suggests that their replacement with dicarboxylato [
170] or bis(2-pyridyl) dioximato groups [
172,
173,
174], respectively, could lead to discrete aggregates (oligomers) rather than polymeric complexes. We give an example in which a bis(2-pyridyl) dioxime provides the inter-SMM linkage.
The ligand of choice was H
2dpd (
Figure 23) and its selection was based on principles of the supramolecular chemistry field [
174]. The molecule can be seen as a fusion of two Hmpko units (
Figure 21, right, with R = Me and R’ = H) at the Me group. The single sp
3 central carbon atom reduces the conformational flexibility, and according to the directional bonding approach of supramolecular chemistry, the combination of a tritopic {Mn
III3} unit with a ~109° ditopic dioximate should give a {Mn
III3}
2 “dimer” with three linkers and parallel {Mn
III3} planes. It was also expected that the coordination by tridentate 2-pyridyloximate groups of three dpd
2− ligands would give a rigidity in the resulting dimeric product, a favorable fact for the retention of the structure in solution. All these design principles turned out to be successful, Equation (23). The I
3− counterions, which are present in the product [Mn
6O
2(O
2CMe)
6(dpd)
3](I
3)
2 (
38), come from I
2 in the reaction mixture, probably from reductive agents, e.g., EtOH, through the 2e
− + 3 I
2 → 2 I
3− process.
At targeted by the selection of H
2dpd, the structure of the hexanuclear dication consists of two {Mn
III3(μ
3-O)}
7+ subunits connected by three 4.111111 dpd
2− ligands (
H in
Figure 5) to give a {Mn
III3} “dimer” of D
3 symmetry; the two {Mn
III3} planes are thus parallel. Each triangular subunit is structurally very similar to that of the “monomer”
37a. Solid-state dc and ac magnetic susceptibility studies show that each {Mn
III3} subunit of the “dimer” is a separate SMM with an
S = 6 ground state and that the two subunits are very weakly ferromagnetically exchange-coupled. Single-crystal high-frequency EPR spectra on
38 display signal splittings suggesting quantum superposition/entanglement of the two SMM subunits. Remarkably, the same spectral behavior is observed in MeCN/toluene (1:1
v/
v) frozen solutions, indicating that the structure of the “dimer” is retained in solution and the weak inter-SMM interaction persists. This work proves that the synthesis of covalently linked oligomers of exchange-coupled SMMs is feasible, with a careful ligand design, and the products can retain their oligomeric nature and inter-SMM quantum mechanical coupling in solution. These results provide scientists with a good background as efforts of using solution methods for deposition of SMMs on surfaces and other substrates continue.

 ,
, 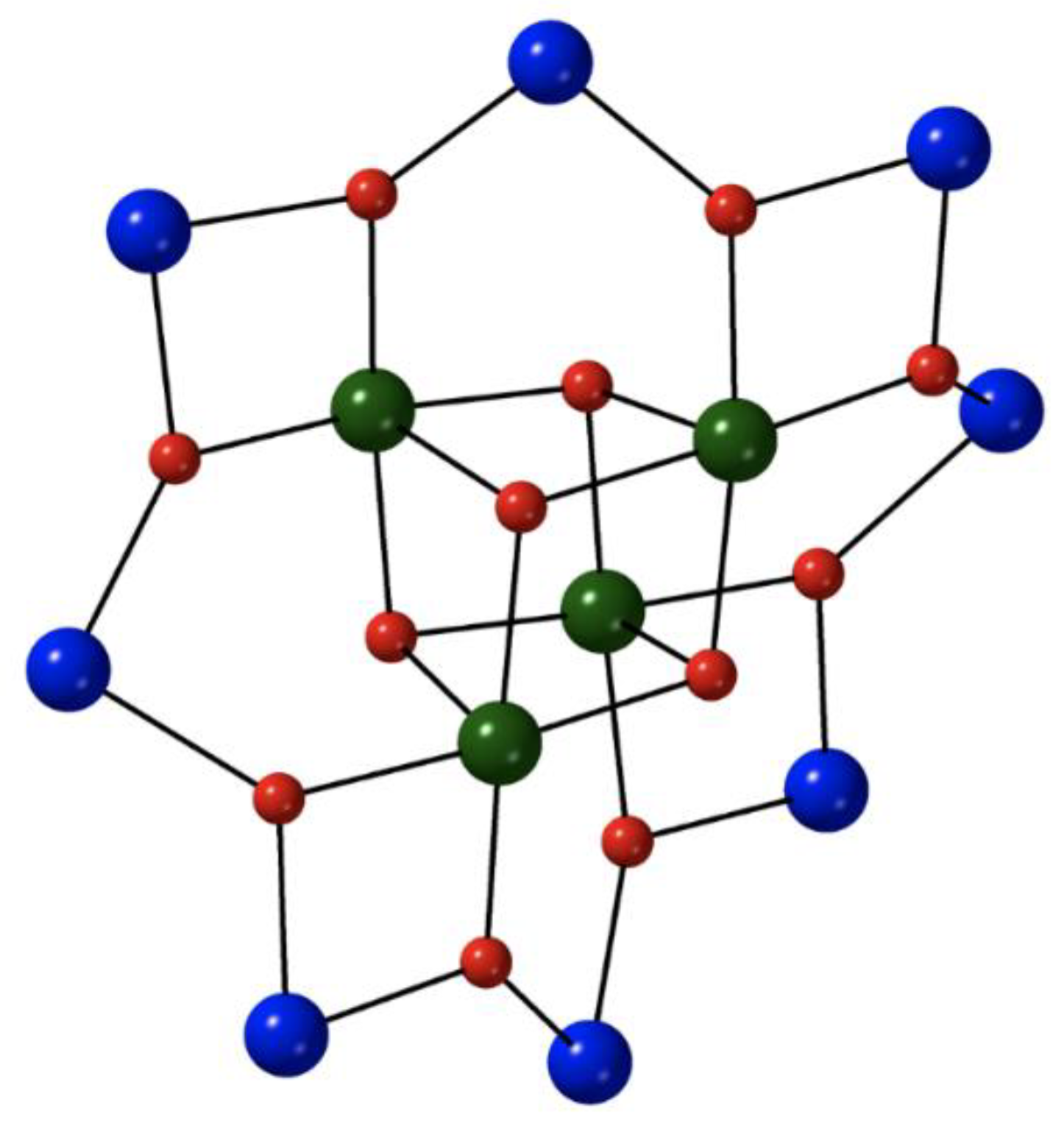{kind=link}
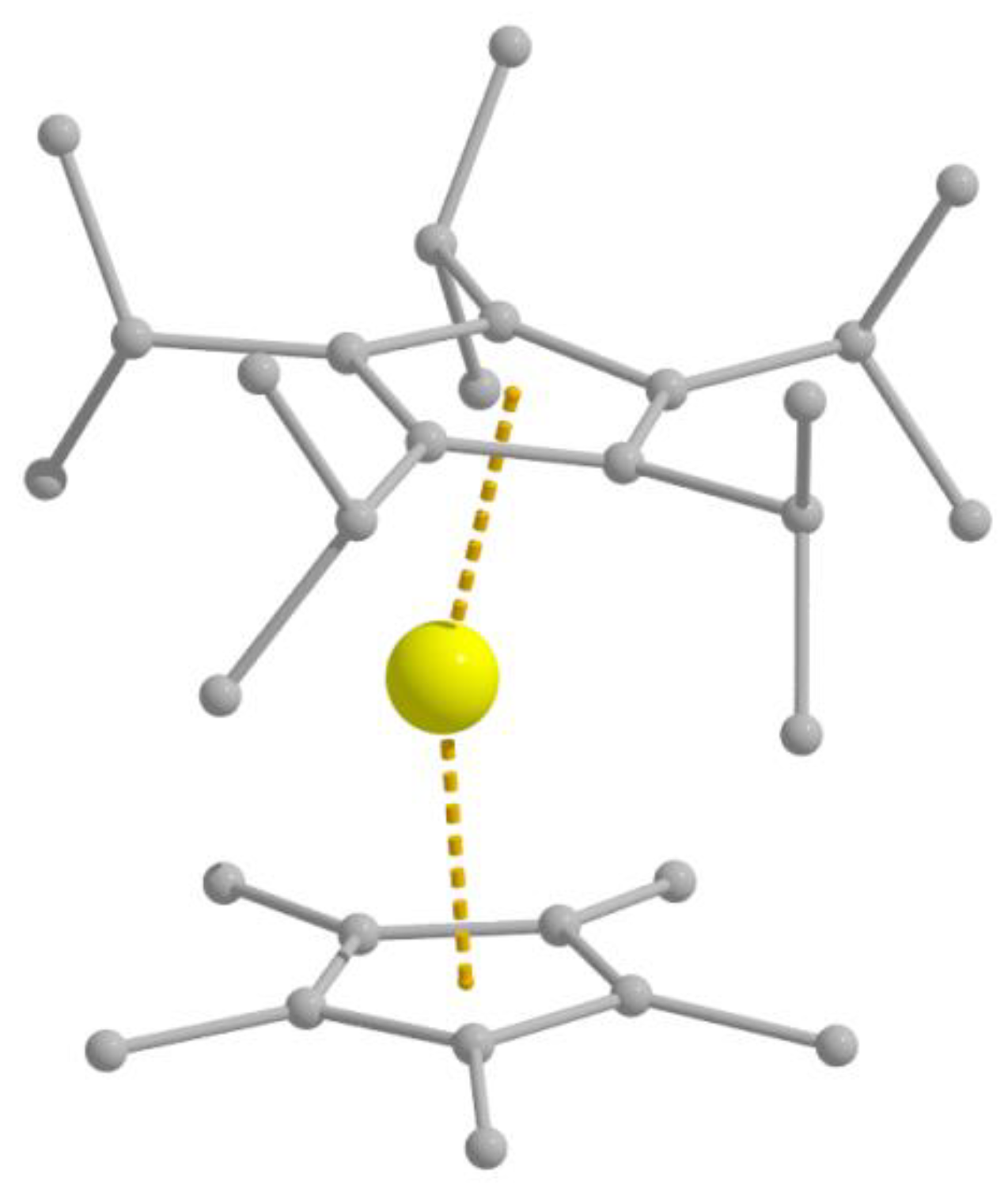{kind=link}
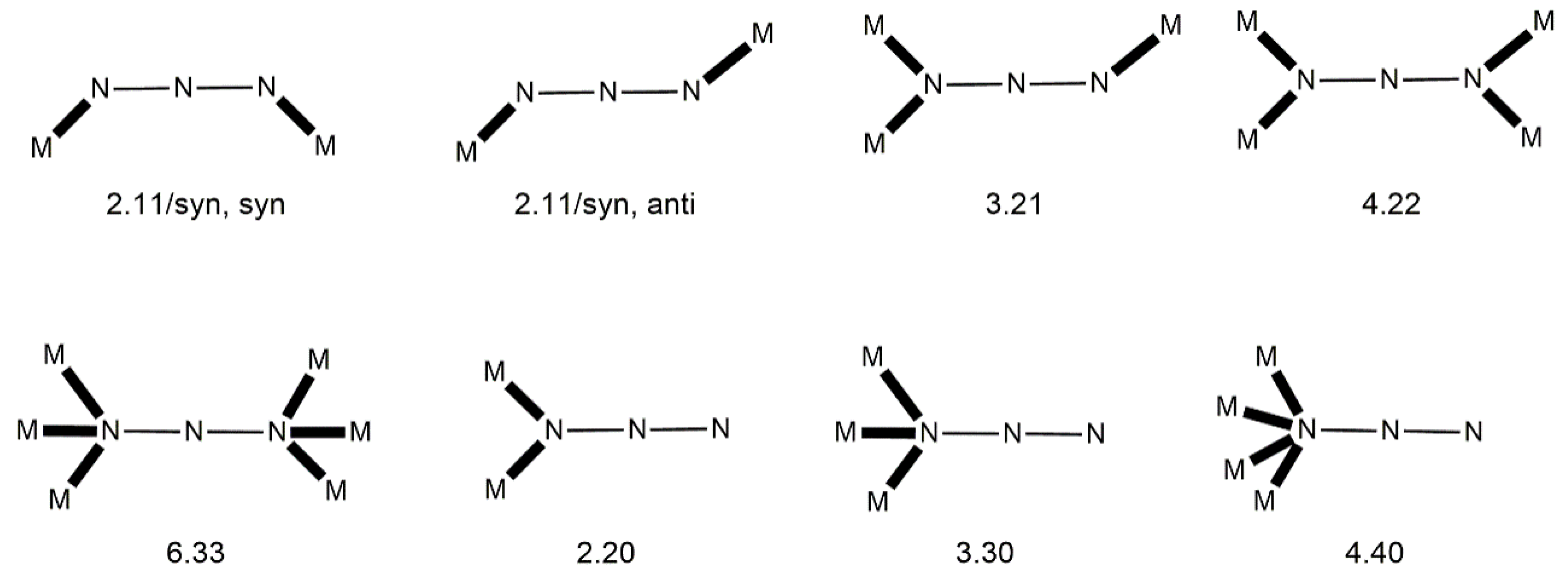{kind=link}
{kind=link}
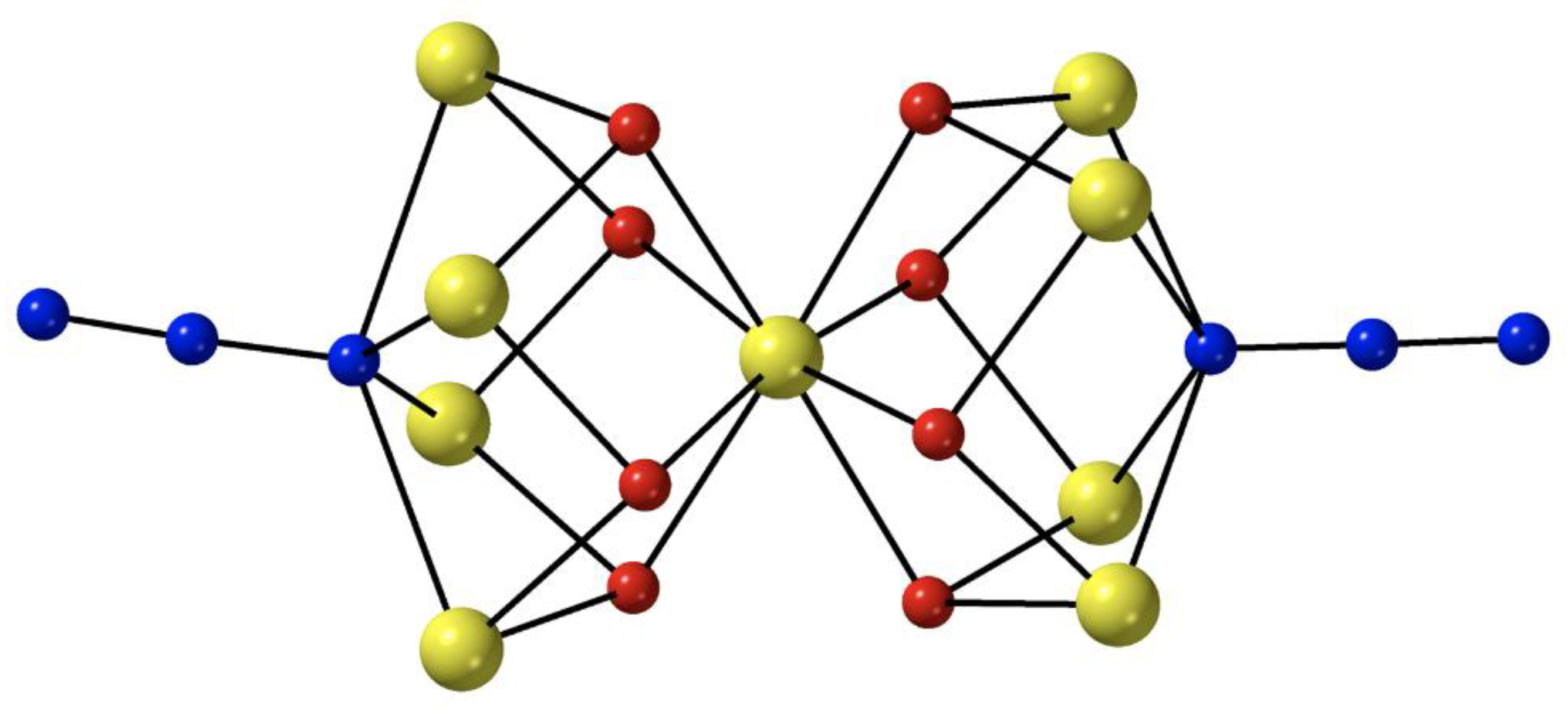{kind=link}
{kind=link}
{kind=link}
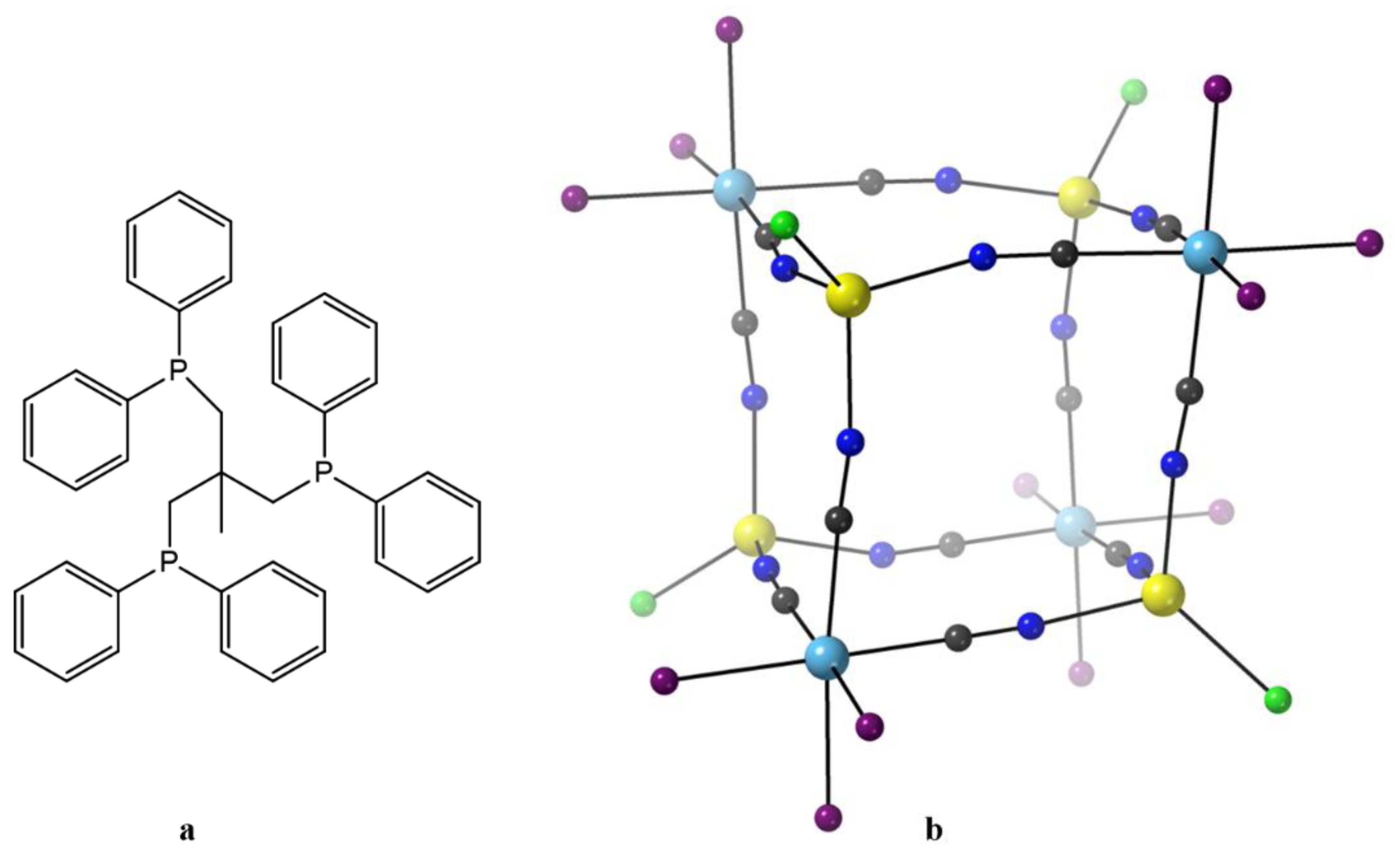{kind=link}
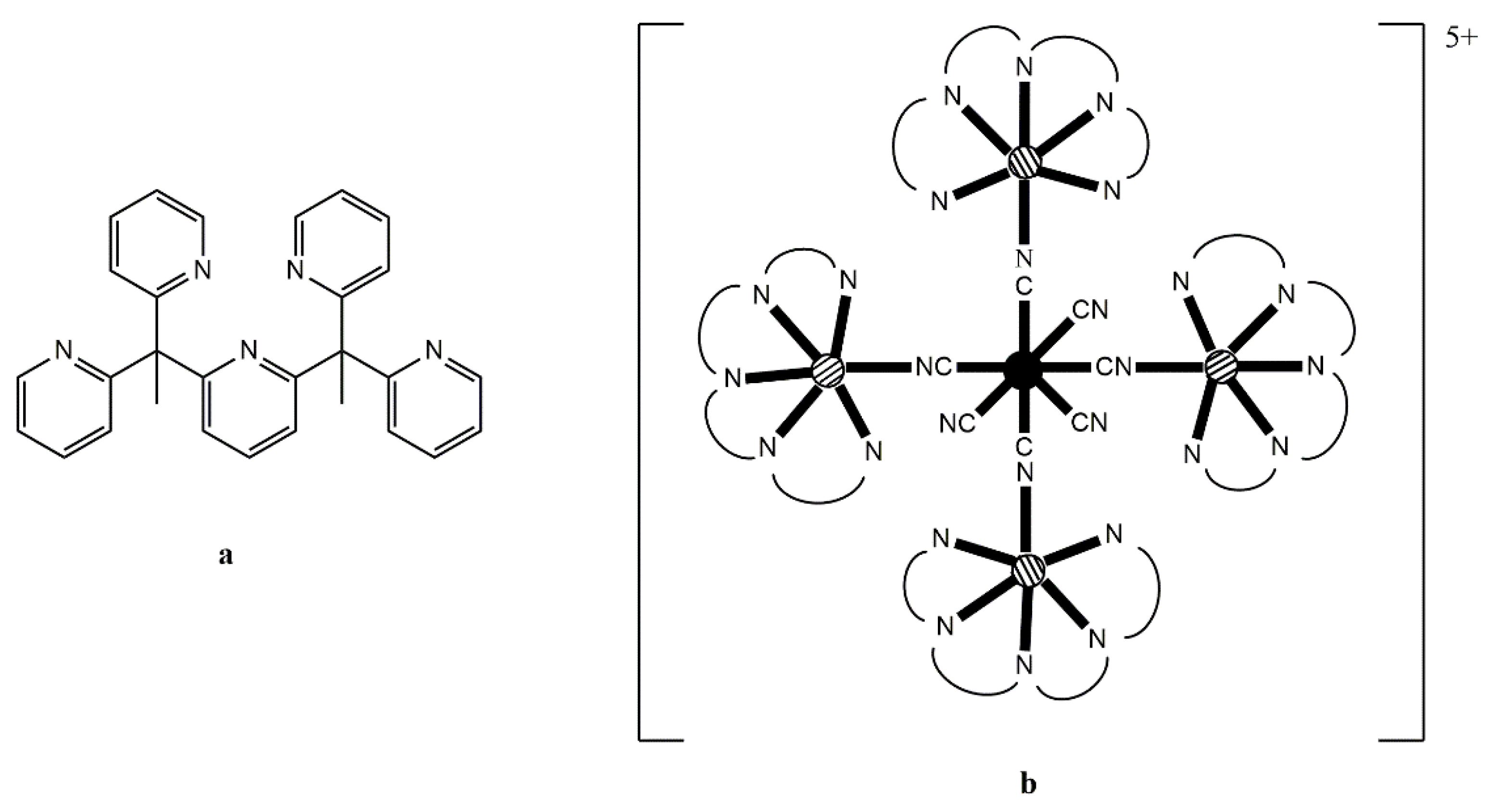{kind=link}
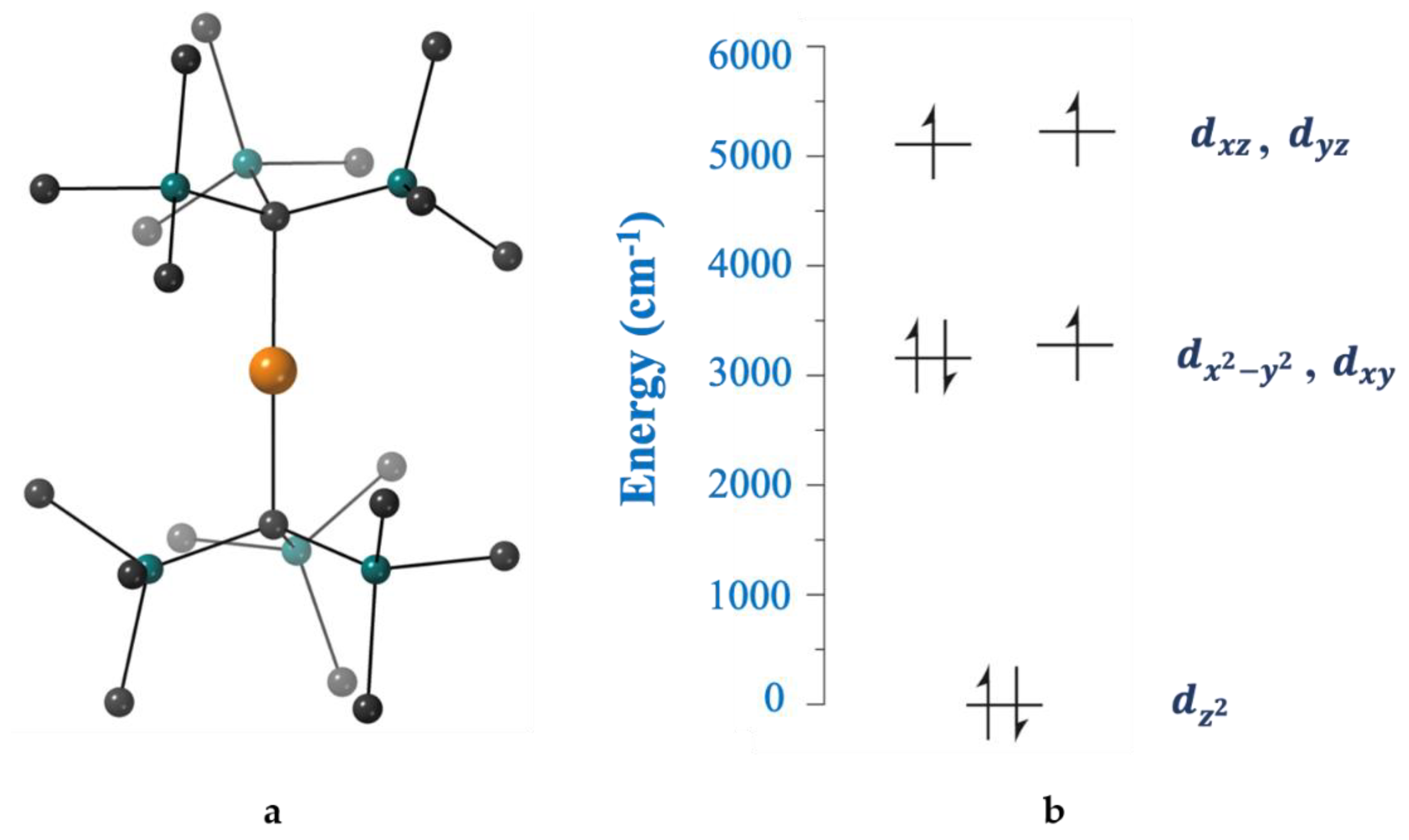{kind=link}
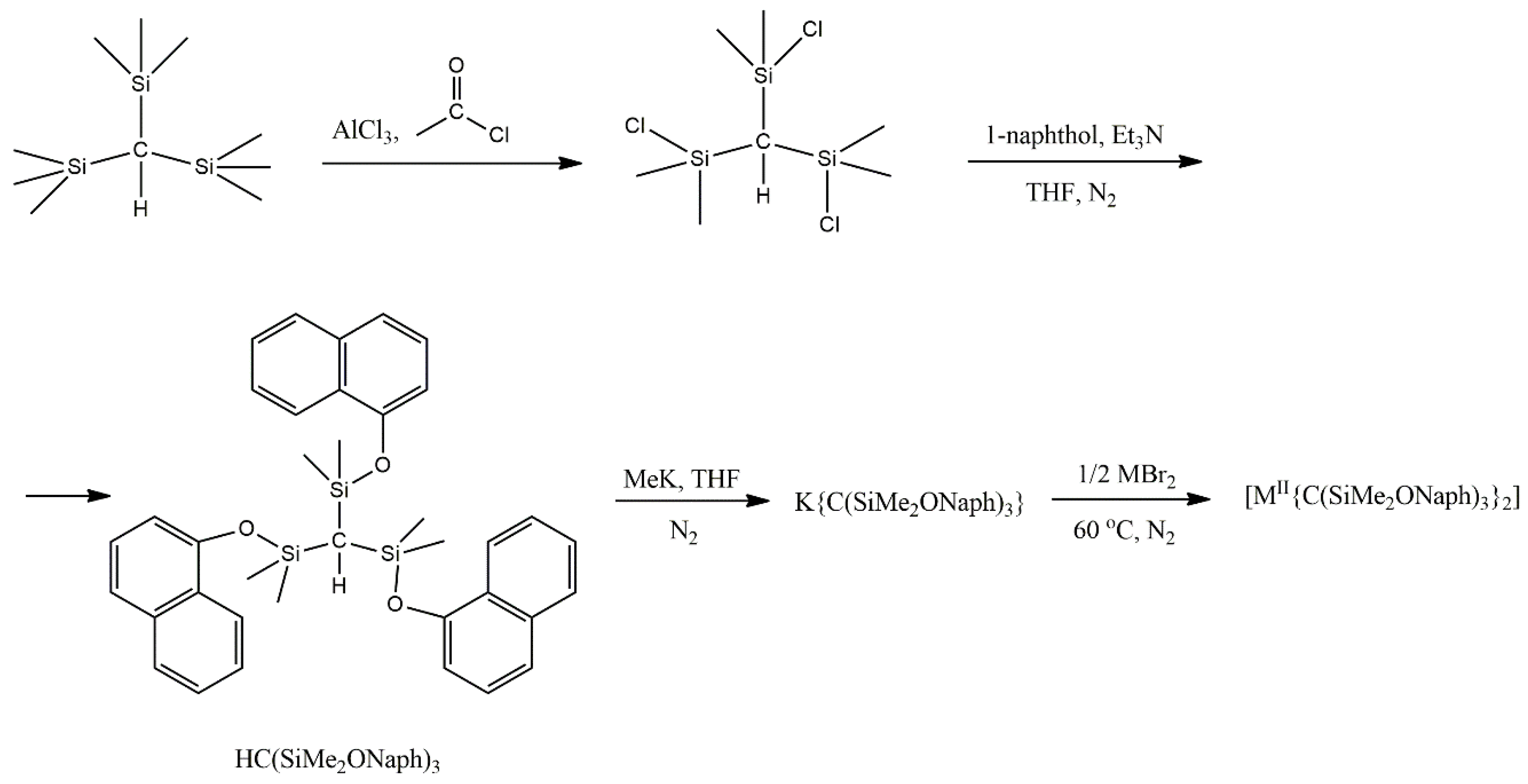{kind=link}
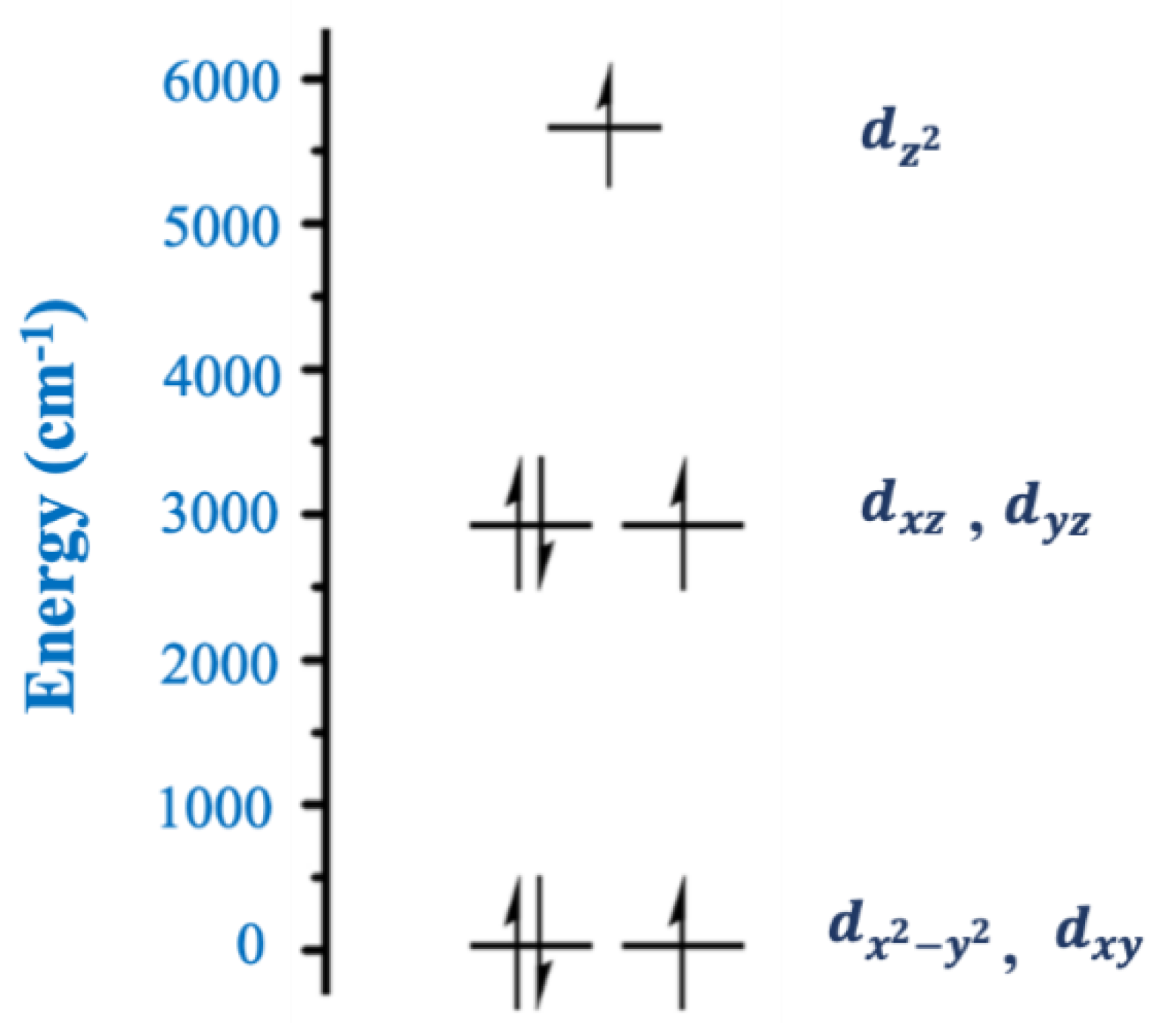{kind=link}
{kind=link}
{kind=link}
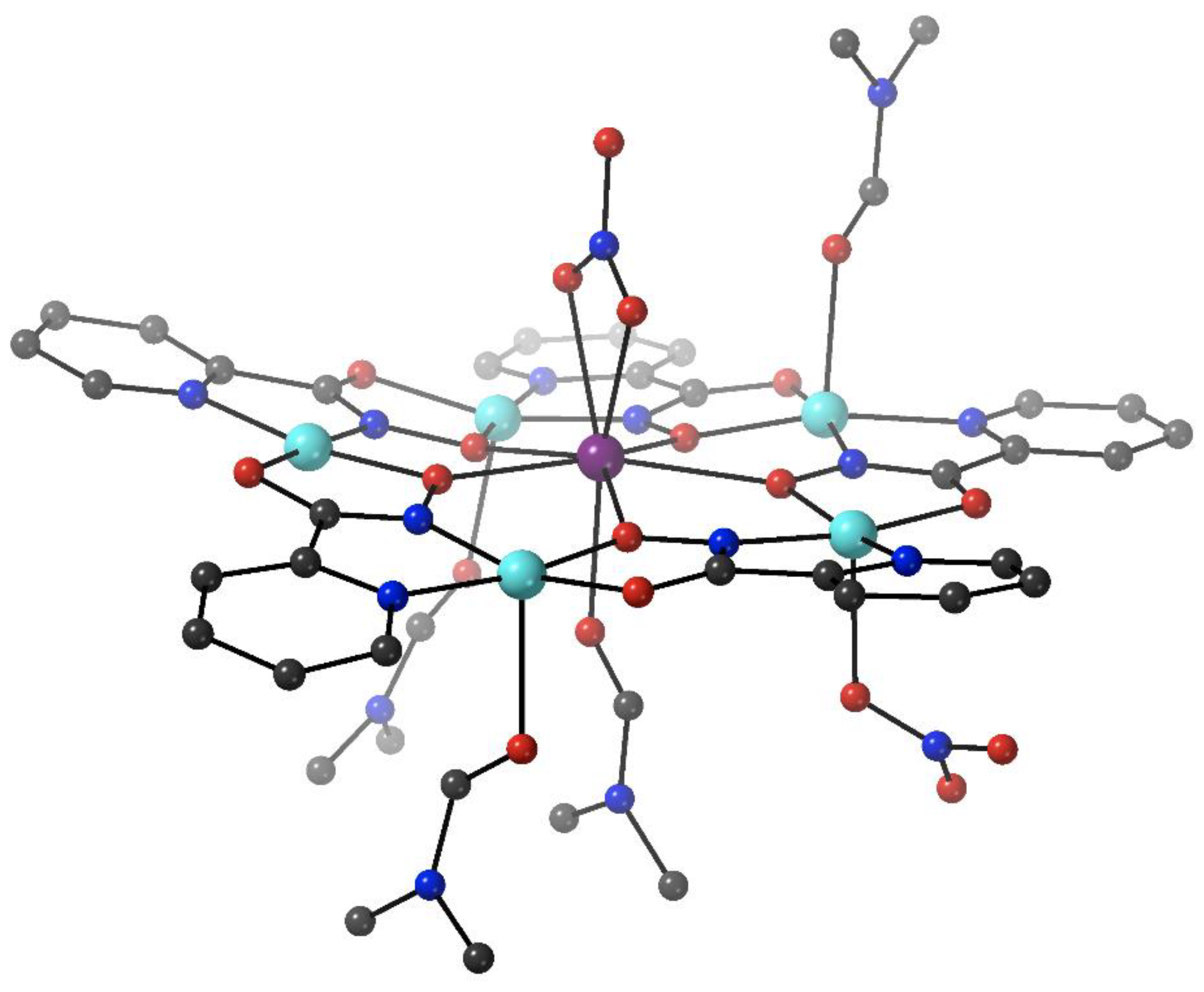{kind=link}
{kind=link}
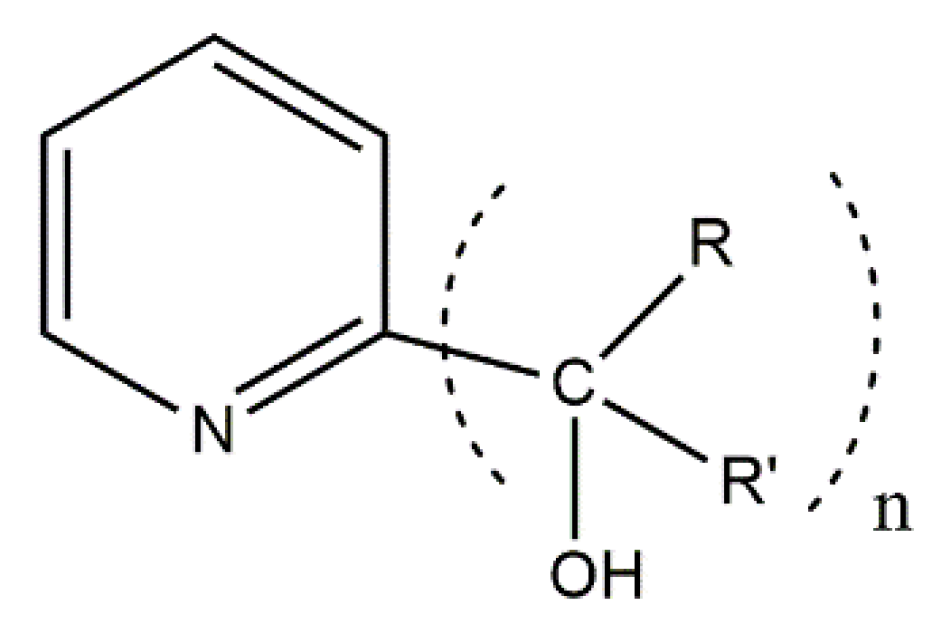{kind=link}
{kind=link}
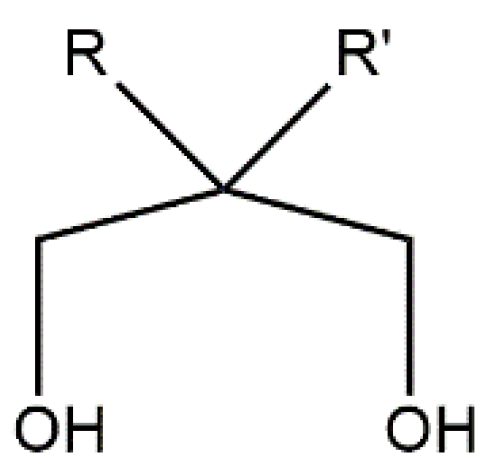{kind=link}
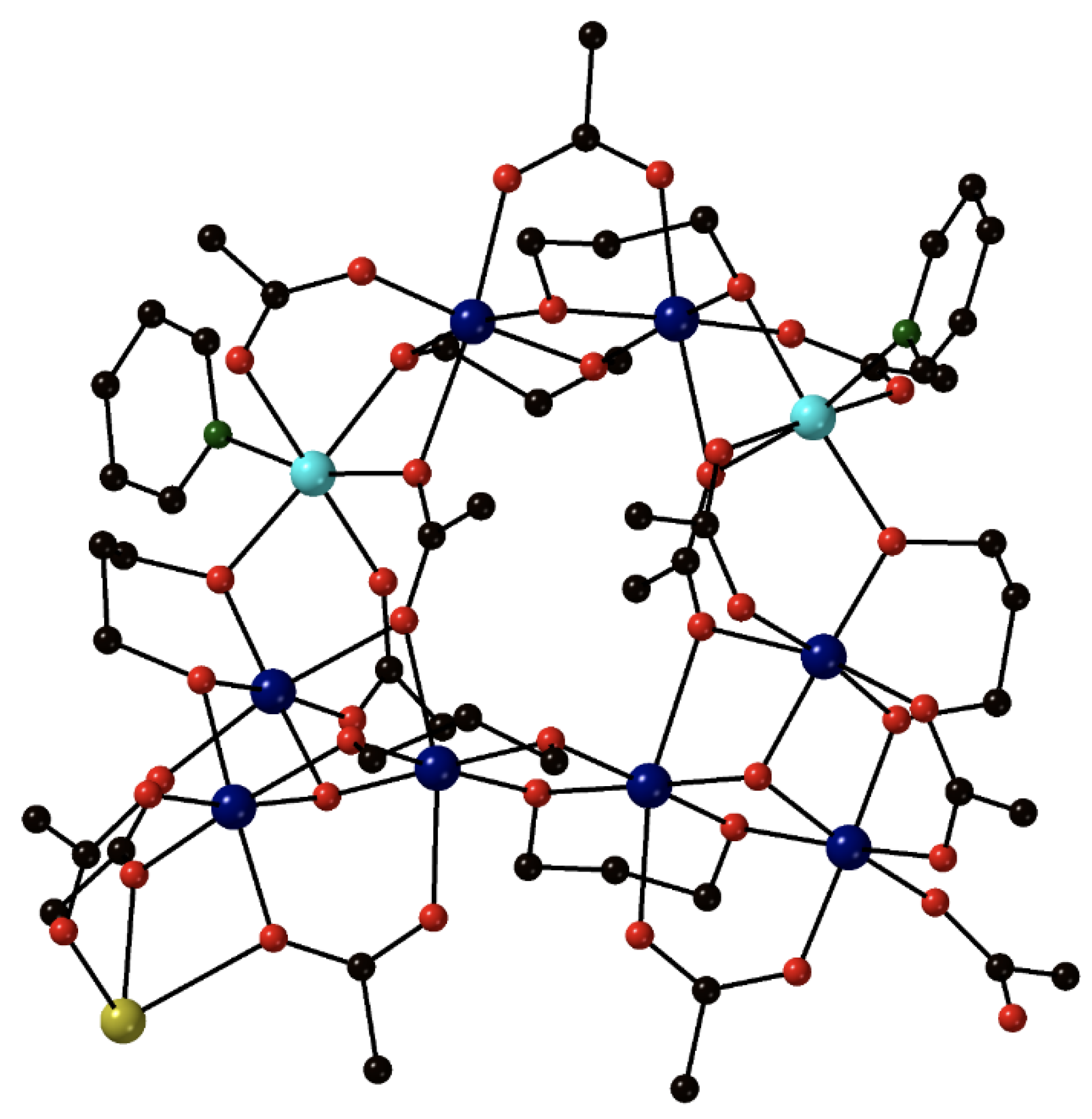{kind=link}
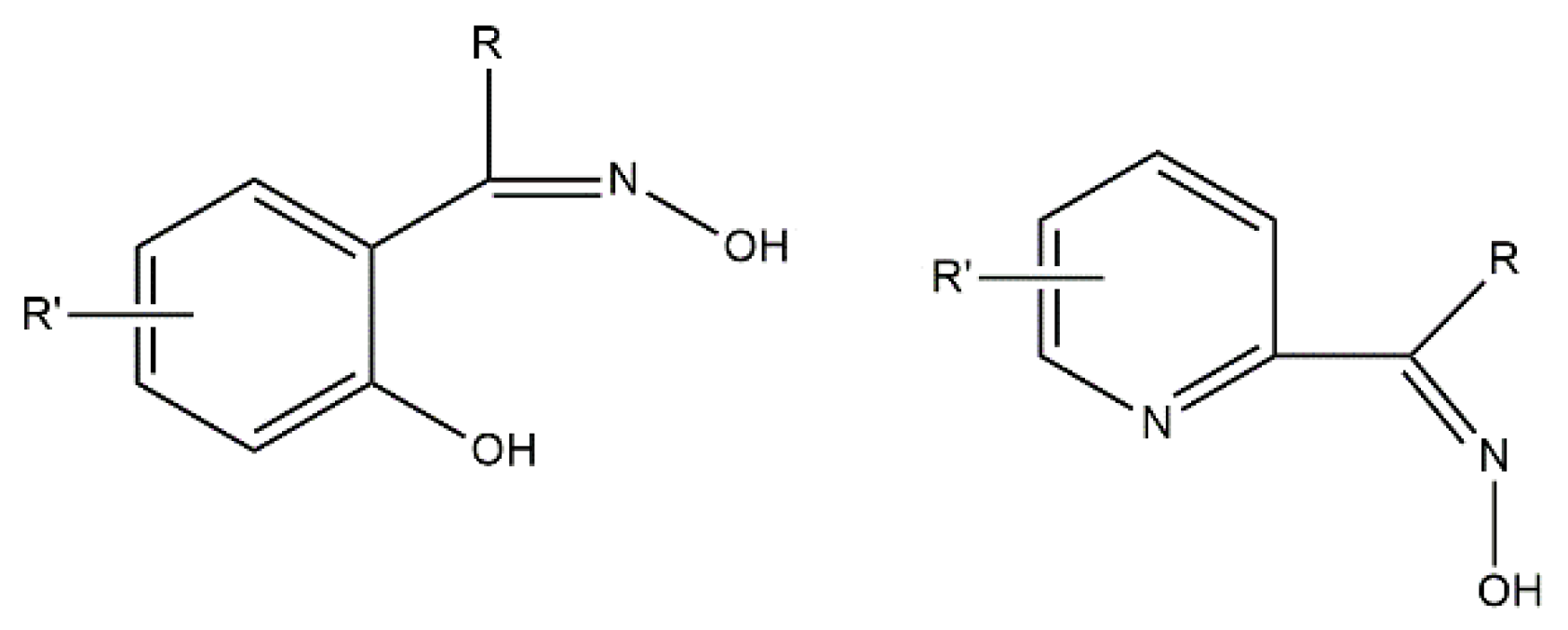{kind=link}
{kind=link}
{kind=link}


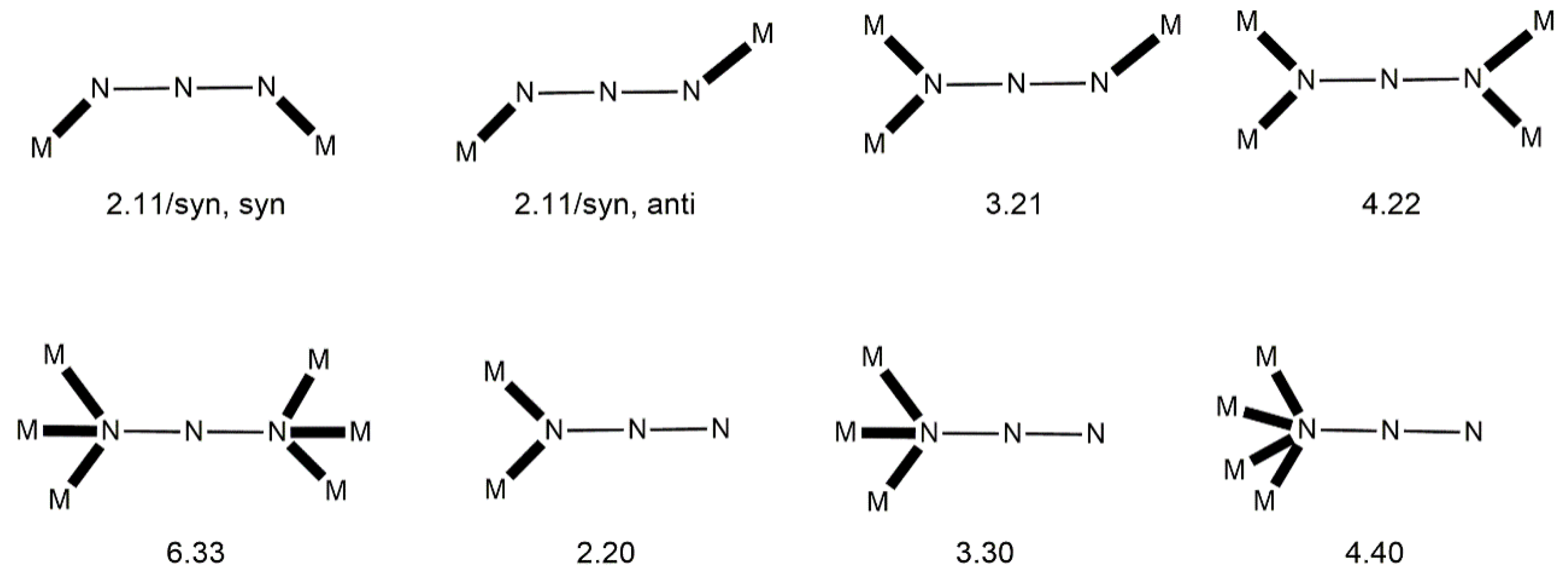

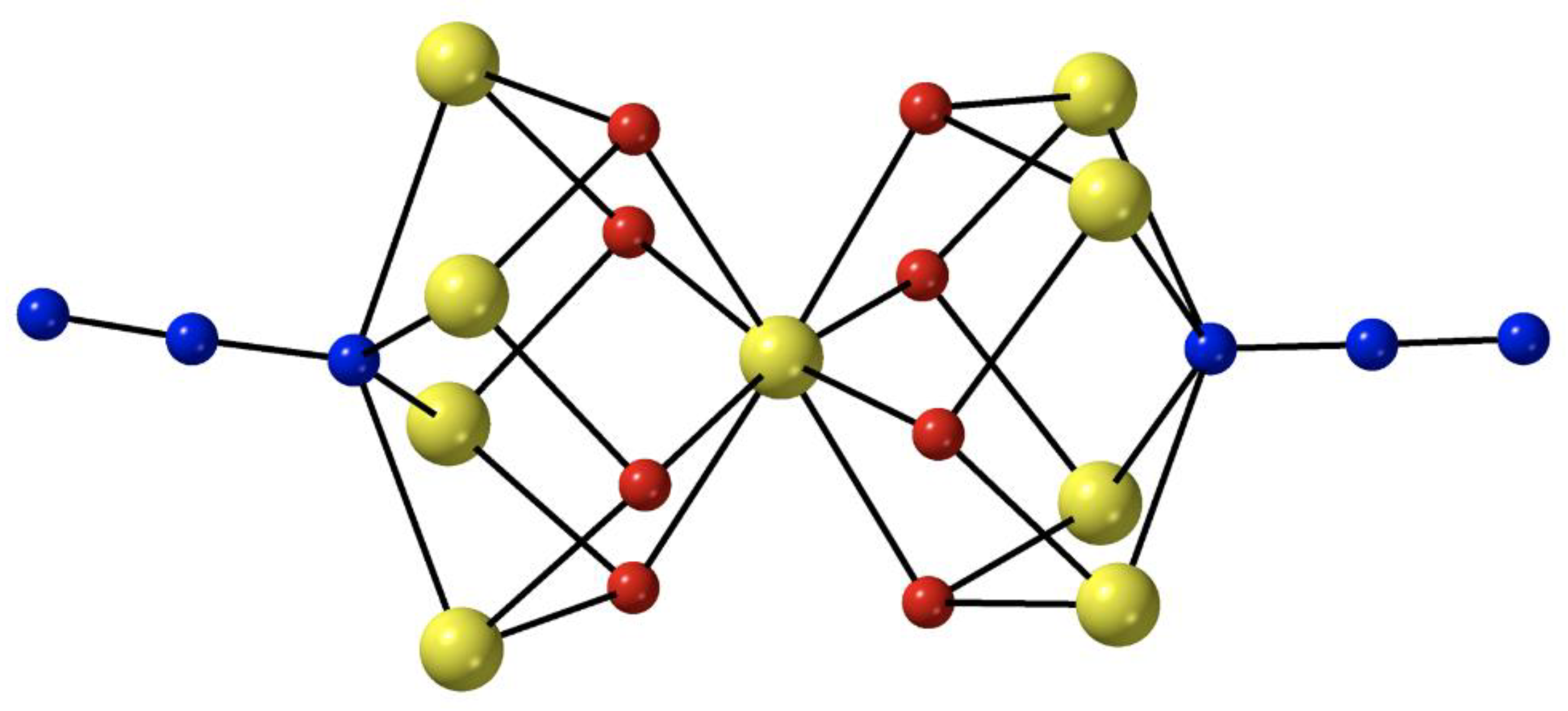
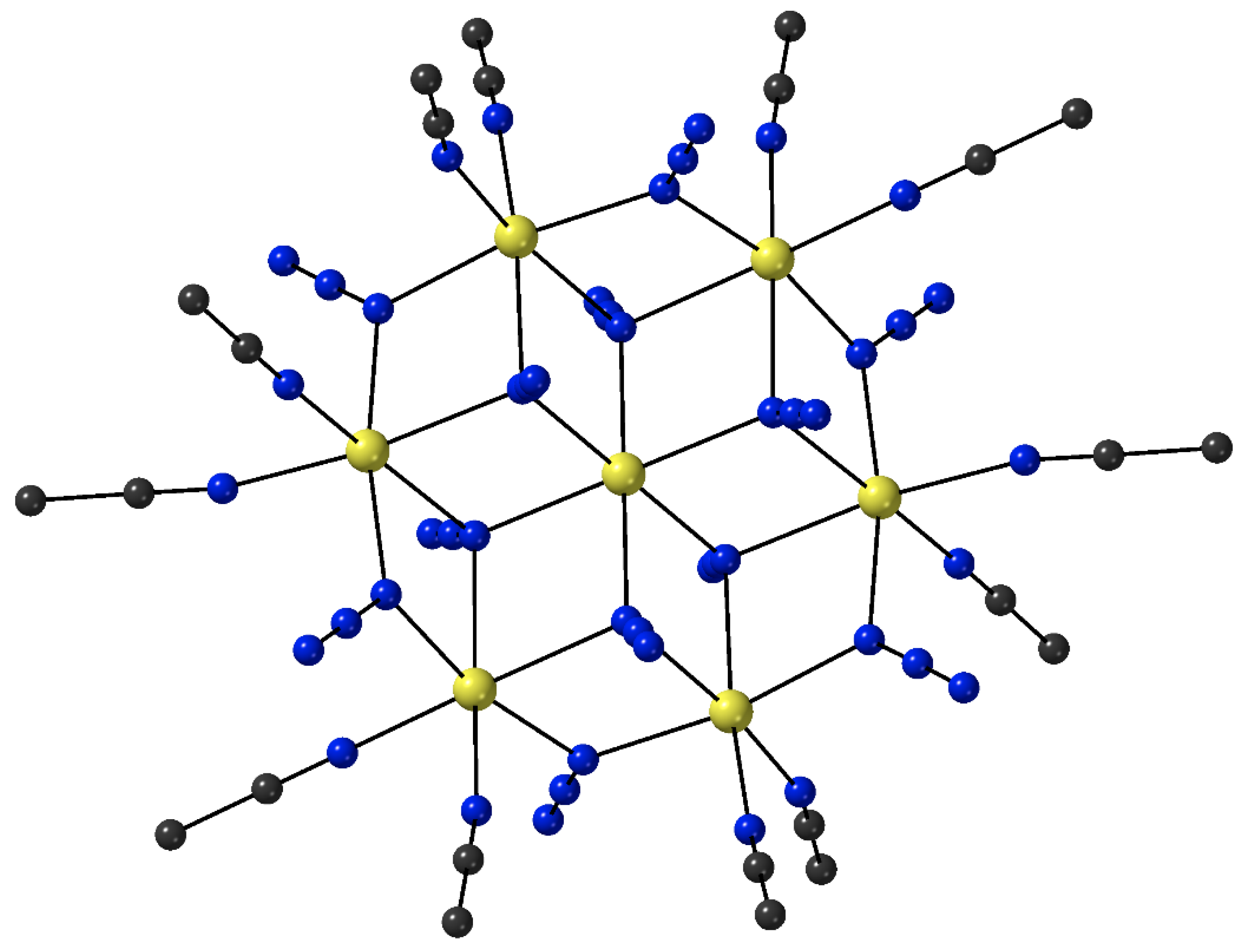
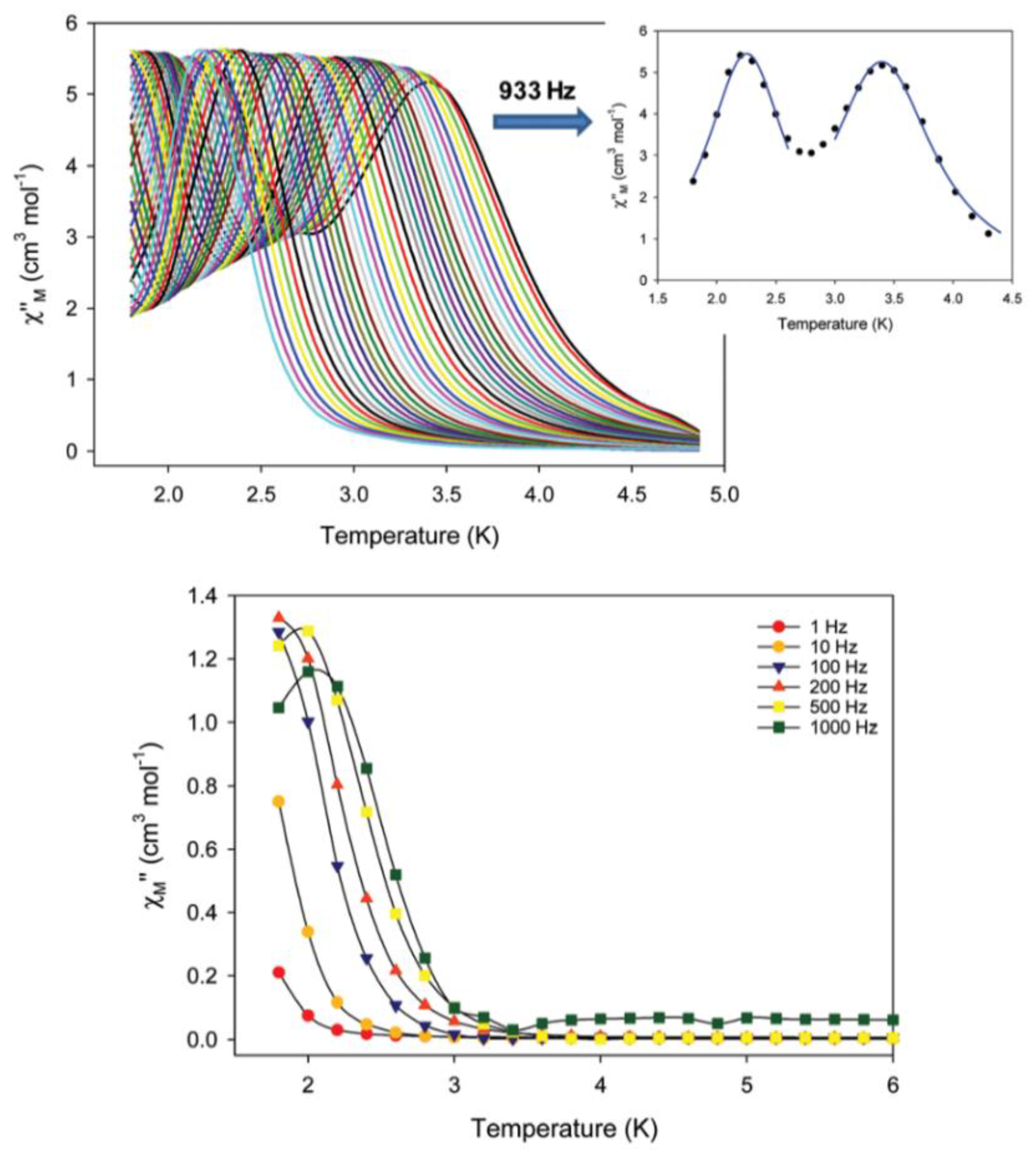
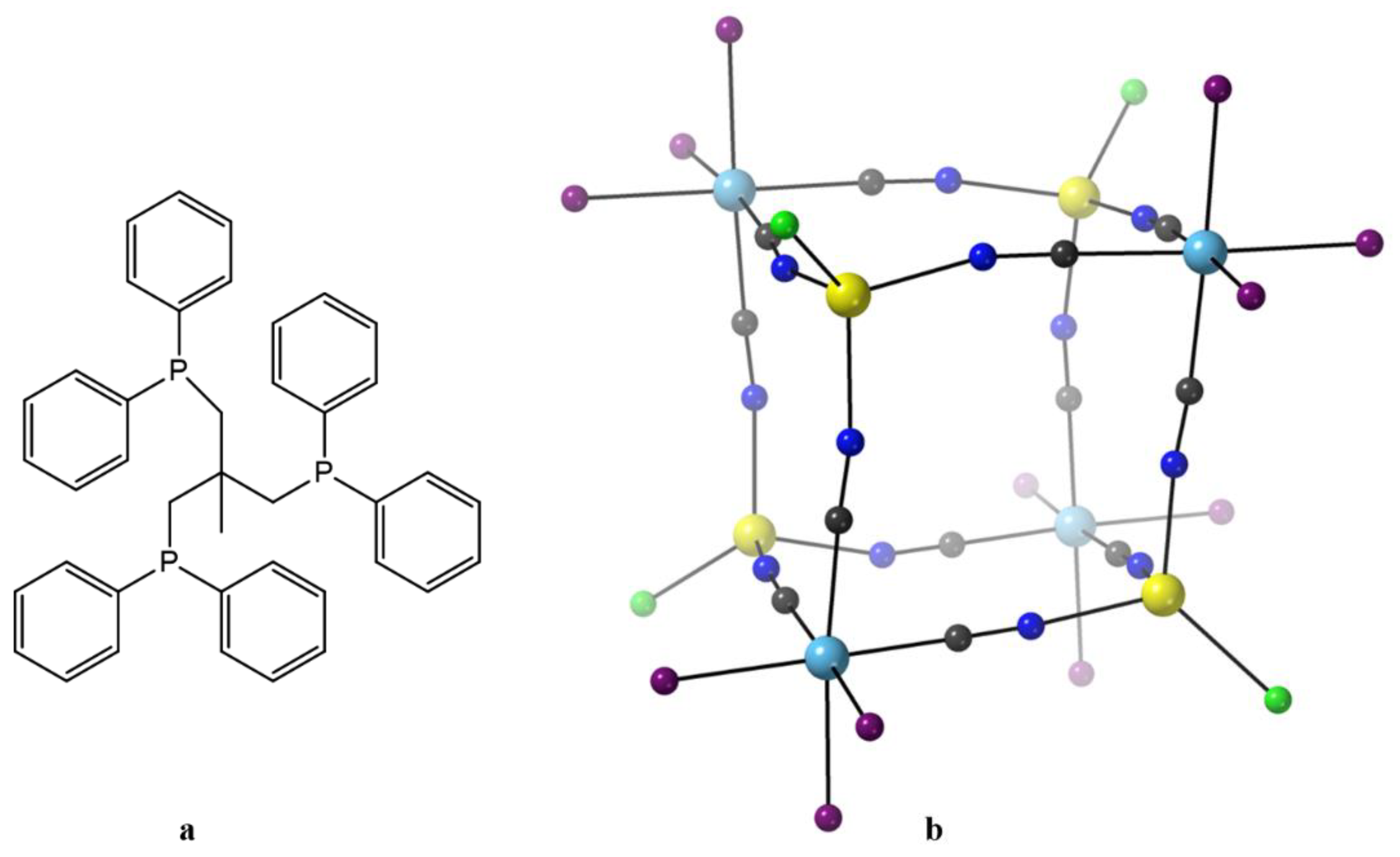
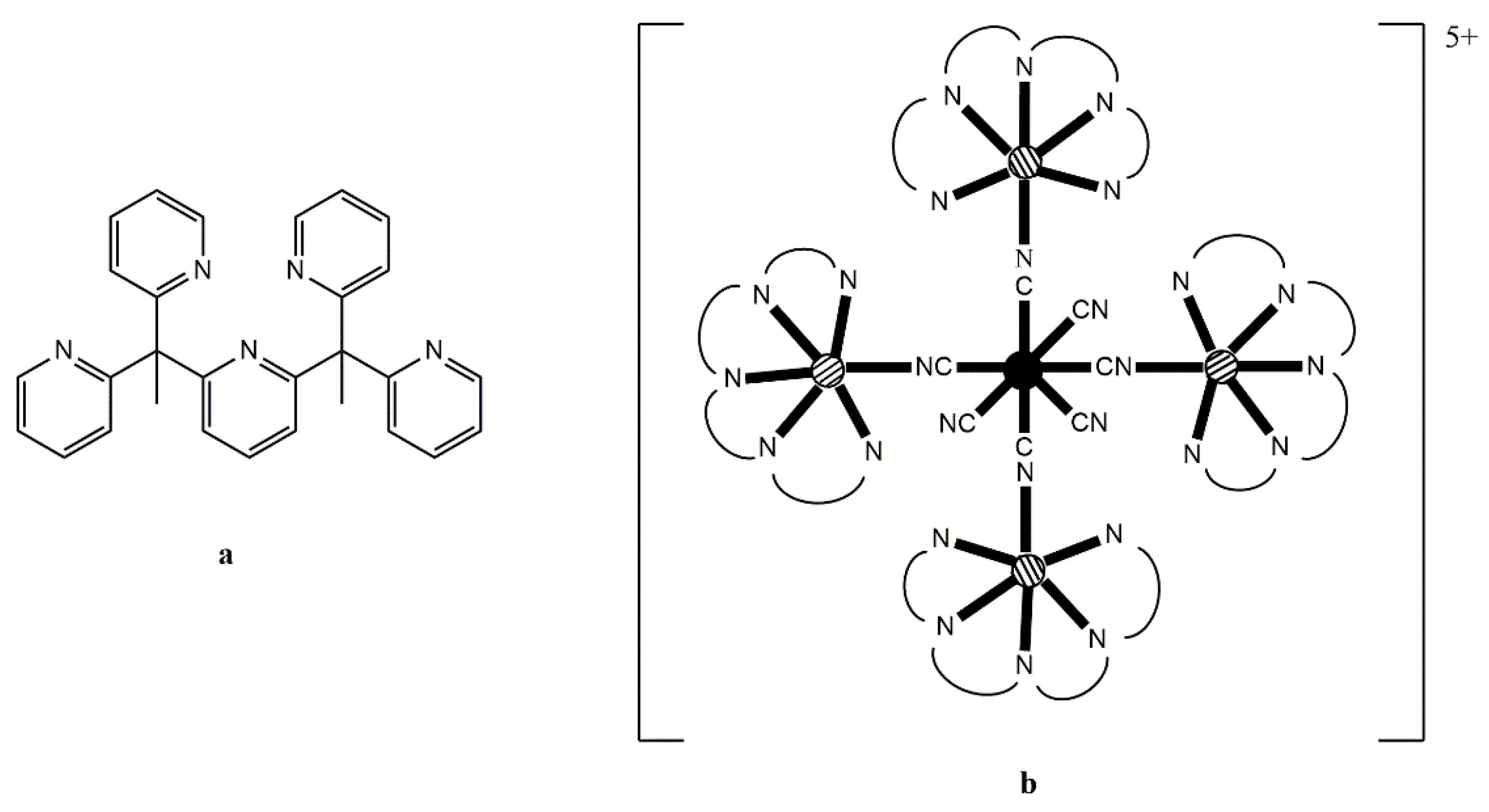

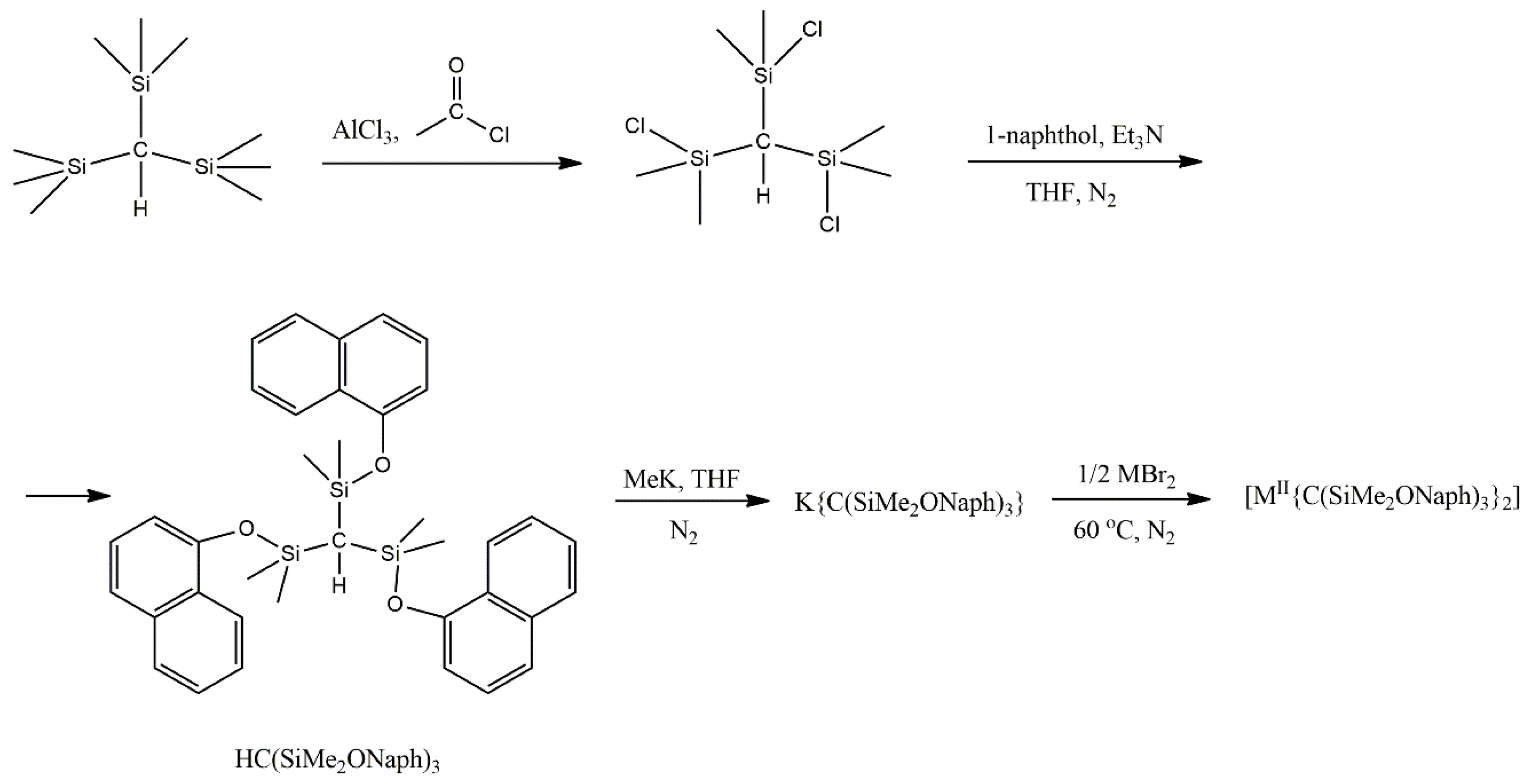
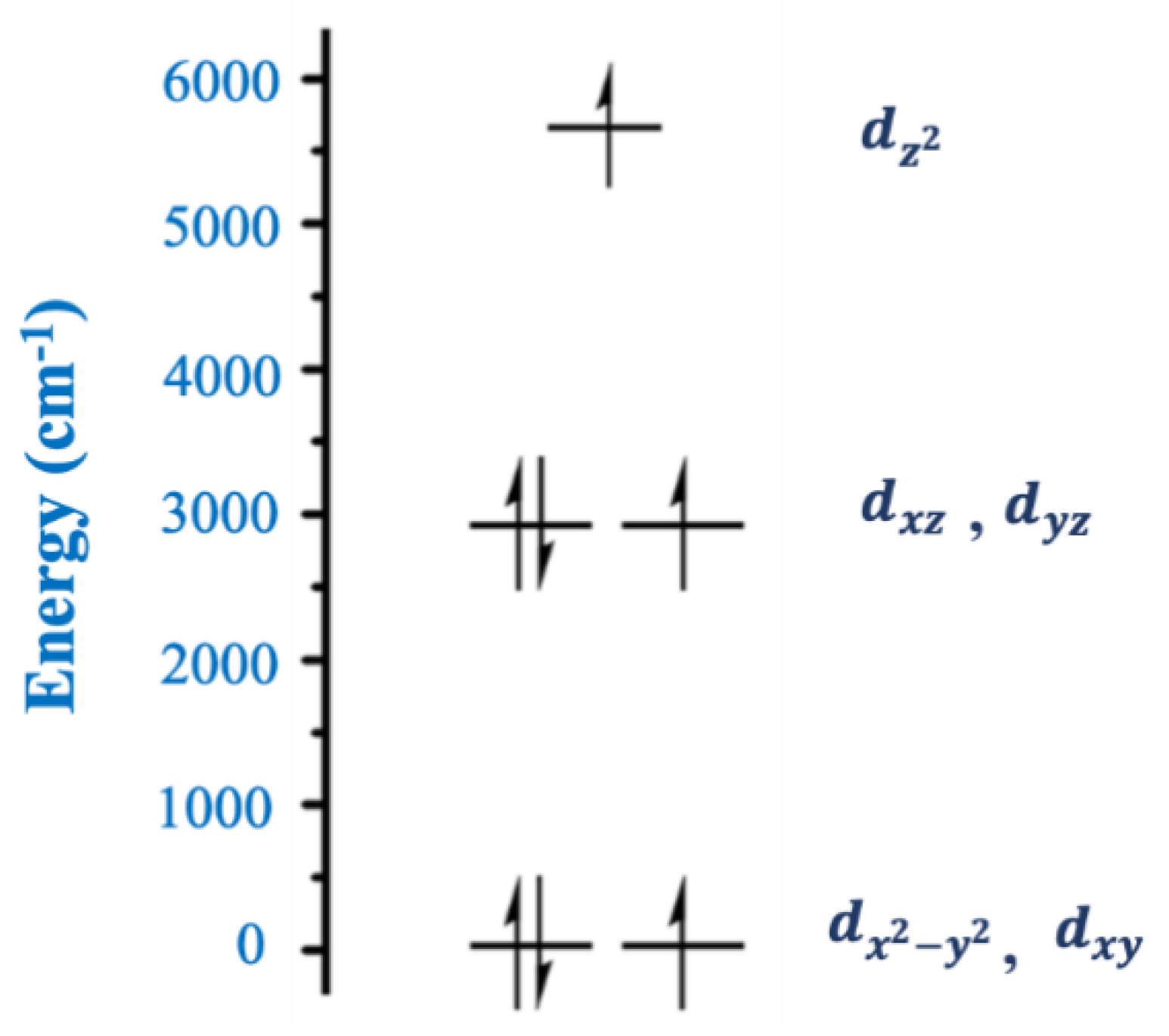

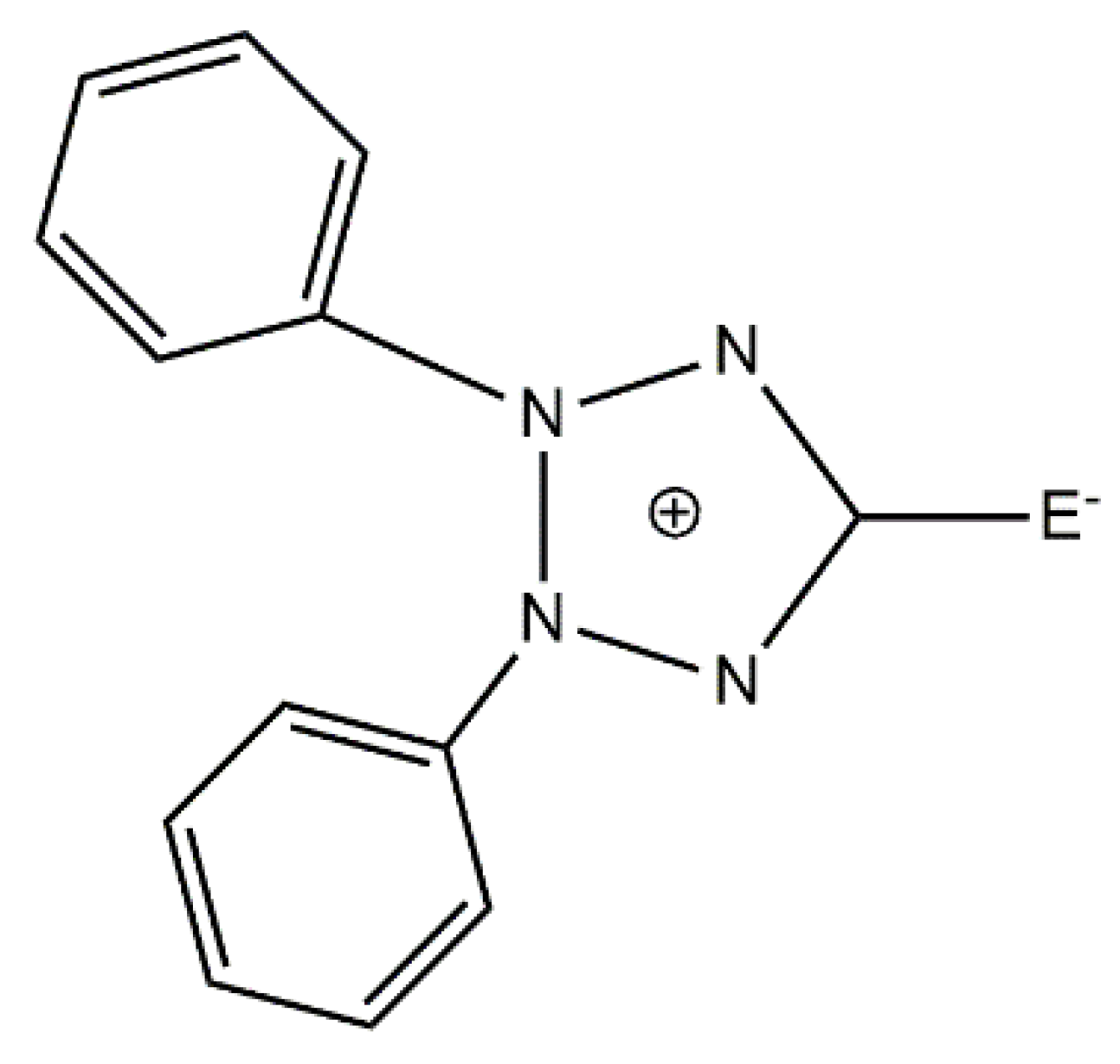




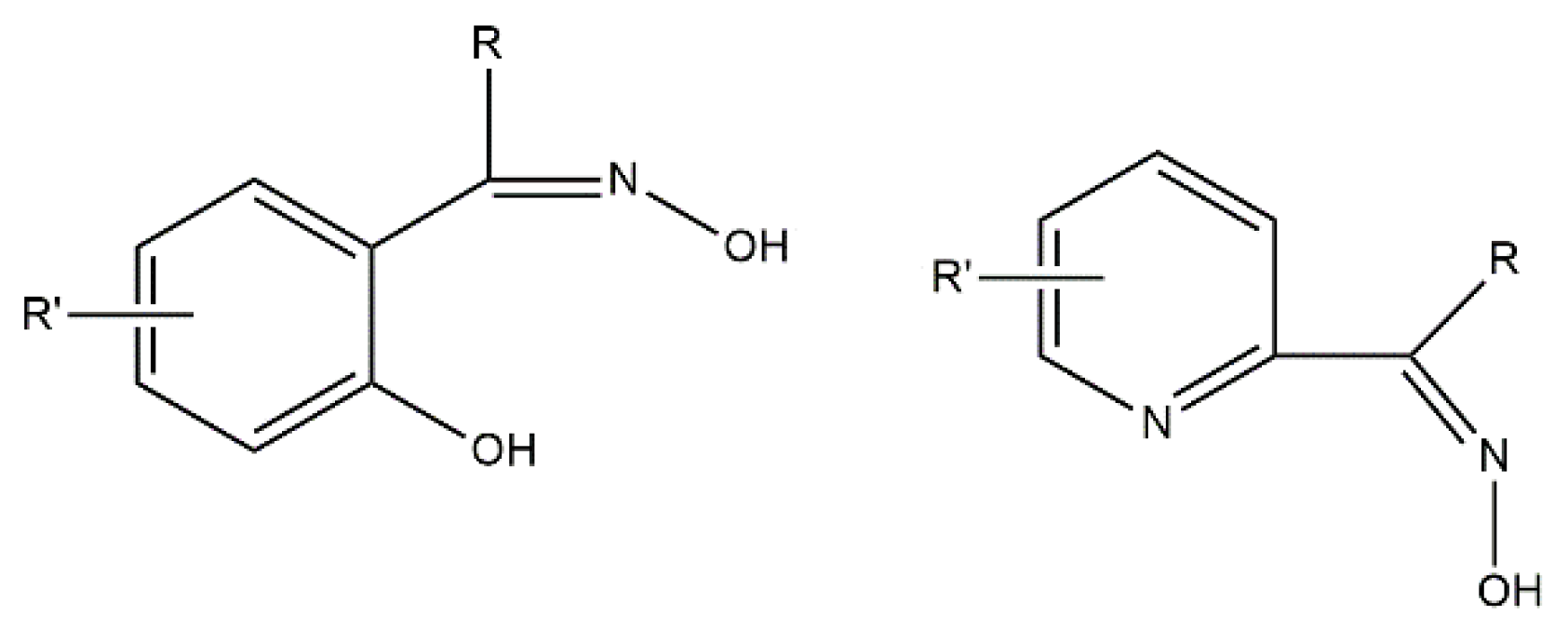
 represent triatomic carboxylate groups. The mixed dashed/solid lines
represent triatomic carboxylate groups. The mixed dashed/solid lines  represent the N,N (small dashed circles), O (small open circle) donor sets of the 2.111 mpko− ligands. The coordination bonds are drawn with bold lines.
represent the N,N (small dashed circles), O (small open circle) donor sets of the 2.111 mpko− ligands. The coordination bonds are drawn with bold lines.