
Unfolded Protein Response and Crohn’s Diseases: A Molecular Mechanism of Wound Healing in the Gut


1
Department of Internal Medicine, Division of Gastroenterology, Hepatology and Nutrition, Virginia Commonwealth University, Richmond, VA 23298-0341, USA
2
Department of Physiology and Biophysics, Medical College of Virginia Campus, Virginia Commonwealth University, Richmond, VA 23298-0341, USA
Gastrointest. Disord. 2021, 3(1), 31-43; https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord3010004
Submission received: 8 January 2021
/
Revised: 2 February 2021
/
Accepted: 5 February 2021
/
Published: 10 February 2021
(This article belongs to the Special Issue Aspects of the Pathogenesis and Management of the Inflammatory Bowel Diseases)
{kind=link}
{kind=link}
Abstract
:Endoplasmic reticulum (ER) stress triggers a series of signaling and transcriptional events termed the unfolded protein response (UPR). Severe ER stress is associated with the development of fibrosis in different organs, including lung, liver, kidney, heart, and intestine. ER stress is an essential response of epithelial and immune cells in the pathogenesis of Inflammatory Bowel Disease (IBD), including Crohn’s disease (CD). Intestinal epithelial cells are susceptible to ER stress-mediated damage due to secretion of a large amount of proteins that are involved in mucosal defense. In other cells, ER stress is linked to myofibroblast activation, extracellular matrix production, macrophage polarization, and immune cell differentiation. This review focuses on the role of the UPR in the pathogenesis in IBD from an immunologic perspective. The roles of macrophage and mesenchymal cells in the UPR from in vitro and in vivo animal models are discussed. The links between ER stress and other signaling pathways, such as senescence and autophagy, are introduced. Recent advances in the understanding of the epigenetic regulation of the UPR signaling are also updated here. The future directions of development of the UPR research and therapeutic strategies to manipulate ER stress levels are also reviewed.
1. Introduction
As an ultimate perinuclear organelle, the endoplasmic reticulum (ER) is a membranous labyrinth network where cell-surface and secreted proteins can be synthesized and maintained with high fidelity through the assistance of molecular chaperones (e.g., glucose-regulated protein 78 kD or immunoglobulin heavy chain-binding protein, GRP78/BiP) and folding enzymes (e.g., protein disulfide isomerases, PDI) [1,2]. Only correctly folded proteins can be transported to the Golgi apparatus. Unfolded or misfolded proteins are retained in the ER and further inversely translocated from the ER lumen to the cytosol by a cellular procedure called the “Endoplasmic-Reticulum-Associated Protein Degradation (ERAD)”. ERAD designates a cellular pathway that targets unfolded or misfolded proteins in the ER for ubiquitination and subsequent degradation, usually by the 26S proteasome [1,2].
An imbalance between the load of misfolded protein generated in the ER and ERAD machinery triggers a series of cytoprotective signaling pathways called the Unfolded Protein Response (UPR) [1,2,3]. Upon the onset of ER stress, GRP78 dissociates from its binding partners: Inositol-requiring enzyme 1α and β (IRE1α and β), activating transcription factor-6α (ATF-6α) and pancreatic ER kinase (PERK). Dissociation of GRP78 from these three complexes activates the protective UPR [1,2,3]. The UPR plays four main functions: (1) Translational attenuation that prevents excessive accumulation of unfolded proteins, (2) up-regulation of ER chaperones and folding enzymes, such as GRP78 and glucose-regulated protein 94 kD (GRP94) are involved in the general folding process to increase the protein folding capacity, (3) enhanced ERAD of unfolded proteins, which strengthen ERAD ability to clear unfolded proteins and send them to the cytoplasm for proteasome-mediated degradation, (4) induction of apoptosis, which happens when the unfolded protein in the ER is overwhelming and the adaptive mechanisms fail to compensate by the first three aforementioned approaches [1,2,3].
The UPR plays an important role in the maintenance of proteostasis by reducing the nascent and misfolded proteins, which are produced under a variety of conditions in physiology and diseases [4,5,6,7,8]. ER stress and activation of the UPR are associated with intestinal epithelial cell damage and apoptosis in CD [5,6,7,8]. UPR-associated genes (e.g., IRE1α, ATF6, and XBP1) have also been implicated in the genetic analysis of CD [5,6,7,8]. A number of studies have shown that ER stress and the UPR play a critical part in shaping immune cell differentiations and functions in order to mount either a protective or a destructive immune response in the host depending upon various conditions [5,8]. Furthermore, intestinal epithelial cells and microbiota contribute to the complexity and dynamic interaction with immune cells within the inflamed gut to resolve the tissue damage, which is induced by a variety of cytokines [6,7]. Therefore, it is understandable that the UPR with its downstream signaling pathways is required in the maintenance of intestinal homeostasis. Moreover, dysfunction of the ER stress response contributes to the pathogenesis of IBD and the complications, such as intestinal fibrosis [9].
In this review, we summarize our current understanding of the role of the UPR involved in the pathogenesis of IBD. Furthermore, we discuss the role of UPR in immunity using macrophages and mesenchymal cells from animal studies as examples. We also emphasize the importance of epigenetic regulation of the UPR and underscore the links between the UPR and senescence, as well as the UPR and autophagy. From a translational perspective, we discuss the possibility of considering the ER stress components as novel pharmacological targets. The review concludes by identifying the future research challenges that need to be addressed to gain a better understanding of the ER and the UPR in physiology and medicine. Figure 1 is a word cloud, which includes all the basic concepts that are discussed further with details in subsequent sections.
2. The Cause of the UPR
In physiological conditions, the ER is responsible for the entry and release of calcium, protein synthesis, and package, lipid metabolism [10]. When cells are subjected to a wide range of stressful conditions, ER stress response reacts with generation of unfolded or misfolded proteins, which triggers UPR to rescue this cellular dysfunction. These stresses commonly include changes in calcium homeostasis, viral or bacterial infection, inflammation, nutrition or energy deficiency, hypoxia, lipid overload, altered redox status, as well as oncogene activation in cancer [7,10]. During UPR, transcription factors such as ATF6 and XBP-1 are activated and translocated to the nucleus to initiate transcription of genes, which are involved in inflammation, cell proliferation, and fibrosis [9,11]. In addition, ER stress plays a role in cellular differentiation, antigen presentation, and stem cell renewal capacity [8]. In 1974, Drs. Claude, Duve, and Palade were awarded Nobel Prize in Physiology or Medicine “for their discoveries concerning the structural and functional organization of the cell", particularly related to the ER using an electronic microscope. ER stress has been studied in both physiologic and pathologic conditions in different organ systems [3,4,5,6,7]. In the gastrointestinal system, it has been studied in epithelial cell differentiation and function, as well as CD during the process of intestinal epithelial cell damage [5,6,7]. The role of ER stress in mesenchymal cells during the development of intestinal fibrosis has been recently reported and is discussed in detail in subsequent sections [9].
3. The UPR in Physiological and Pathological Conditions (Particularly, IBD)
The UPR detects alterations in the balance of protein folding burden and capacity within the ER. The three main sensors of ER stress, including IRE1, PERK, and ATF6, work together to restore proteostasis by modulating transcription, translation, and mRNA decay [1,2,3]. It should be noted that there are recent, more comprehensive reviews on the role of ER stress and UPR in cancer, kidney disease, metabolic disease, and other autoimmune diseases that can be accessed for further details [3,4,12].
UPR and IBD
Genome-wide association studies (GWAS) have identified more than 240 susceptibility loci in patients with IBD [13,14]. IBD is a lifelong immune response dysregulated in the gastrointestinal tract and affects up to 1 in 200 people in developed countries [15]. IBD has two major forms of diseases: CD and ulcerative colitis, with distinctive features in terms of clinical demonstration and pathogenesis [16]. CD is considered as an immune response-mediated, chronic remittent and relapsing intestinal disorder featured with transmural inflammation in a genetically susceptible patient [17]. Ulcerative colitis is more common worldwide than CD. Colonic epithelial cells (colonocytes), mucous barrier abnormality, and epithelial barrier defects are strongly implicated in the pathogenesis of ulcerative colitis [18].
Several susceptibility loci encode proteins with important roles in proteostasis. Three ER relevant genes are identified that include Orosmucoid-like 3 (ORMDL3) [19,20], anterior gradient 2 (AGR2) [21,22], and XBP1 [23]. ORMDL3 is a key UPR inducer by affecting calcium homeostasis in the ER and a risk locus for both CD and ulcerative colitis [13,14]. Moreover, it selectively activated the ATF6 arm of the UPR in lung epithelia and induced the expression of SERCA2B, also known as ATP2A2, which might be involved in airway remodeling [19]. However, the role of ORMDL3 in IBD is not known yet. ORMDL3 polymorphisms variant was reported to be associated with susceptibility to ulcerative colitis (UC) in the Lithuanian early-onset IBD population [20].
The protein disulfide isomerase AGR2 is highly expressed in secretory cells, such as Paneth and goblet cells, with the highest levels in the ileum and colon [20]. The genes encoding for the human homologs AGR2 are localized on chromosome 7p21.3, which is a susceptibility region for IBD supported by linkage analyses [21]. Maurel et al. [22] showed that AGR2 dimers acted as sensors of ER homeostasis and were disrupted upon ER stress to promote the secretion of AGR2 monomers. ER proteostasis-mediated control of AGR2 dimerization, which might depend on TMED2, promoted AGR2 release in the extracellular environment, thereby, enhancing monocyte recruitment and pro-inflammatory phenotypes. Early reports showed that loss of XBP1 in the intestinal epithelial cells using XBP1-/- mice caused progressive Paneth cell death and spontaneous inflammation in mouse ileum [23].
ER stress can also be blocked with anti-inflammatory treatment, which indicates a link between the UPR and inflammation. In intestinal epithelial cells, which were isolated and cultured from inflamed IL10-/- mice as well as patients with IBD, an increase in GRP78 expression under chronic inflammation can be completely blocked by Grp78 knockdown, or by adding IL-10 to TNF-stimulated IL-10 receptor-overexpressed epithelial cells [24]. The anti-ER stress effect of IL-10 was partially due to IL-10-induced p38 activation, blockage of nuclear translocation and recruitment of ATF6 to the Grp78 promoter [24]. This study suggests that in the absence of an anti-inflammatory cytokine in the epithelium, dysregulation of ER stress may contribute to chronic inflammation-induced intestinal epithelial damage.
During chronic ER stress, the UPR induces a series of adaptive cellular events to maintain a proper proteostasis in order to restore the regular cellular functions, which include glycosylation for protein folding, oxidative stress, calcium translocation, and autophagy [25]. Activation of components in the innate and adaptive immune responses plays an important role in the development of chronic intestinal inflammation [5,8]. Although the loss of eIF2α-phosphorylation did not affect the normal IEC proliferation or differentiation in AAIEC mice, which expressed non-phosphorylatable Ser51Ala mutant eIF2α in IECs, these mice showed defective UPR gene expression and were more susceptible to dextran sulfate sodium (DDS)-induced colitis, suggesting the physiological importance of epithelial eIF2a-P in mucosal homeostasis [26]. AA IECs exhibited a defective UPR signaling and ER-associated mRNA translation, which may contribute to Paneth cell dysfunction under normal conditions [26]. Severe inflammation was also found in the ATF6α-/-mice and the chaperone protein p58IPK-/- mice as well as in the IL-10-/- mice [27]. Taken together, these data showed the complexity of interactions between ER stress, inflammation, and immunity.
4. The UPR in Macrophage and Mesenchymal Cells during the Immune Response
ER stress and immunity are usually intertwined together during different stages of the inflammatory process in a variety of human diseases [5,8].
In C/EBP homologous protein (CHOP)-/- mice, bleomycin-induced lung fibrosis was significantly attenuated compared to wild type mice [28], while administration of tauroursodeoxycholic acid (TUDCA), a chemical chaperone, inhibited bleomycin-induced inflammation and fibrosis in mice [28]. Endo et al. [29] showed that LPS-induced inflammation in the lung of CHOP-/- mice was also attenuated, in addition to the decrease in neutrophil infiltration, IL-1β, and caspase-11 expression. However, Ayaub et al. [30] showed that ECM deposition was increased with the proliferation of arginase-1-positive lung macrophages in CHOP-/- mice. Paradoxically, GRP78+/- haplo-insufficiency mice were significantly protected against bleomycin-induced lung fibrosis due to a decrease in the population of lung macrophages with positive stain for cleaved caspase-3. These data suggest that GRP78- and CHOP-mediated macrophage apoptosis may have opposite roles in response to bleomycin-induced fibrosis.
In a mouse model of nonalcoholic steatohepatitis, CHOP-/- mice demonstrated severe liver damage, inflammation, and fibrosis compared to CHOP wild type due to the increase in activated macrophages. Persistence of net accumulation of these activated macrophages in the liver potentiated liver steatohepatitis in CHOP-/- mice [31]. In another study, Yao et al. [32] reported that CHOP-/- diminished alternatively-activated-macrophage phenotype (M2) and reduced M2 filtration in the mouse lung after bleomycin treatment. Activated M2 macrophages secreted TGF-β and plate-derived-growth-factor (PDGF) to induce activation of myofibroblasts and led to tissue fibrosis. Taken together, the role of CHOP and GRP78 during ER stress should be examined and interpreted carefully because they may have opposite effects on macrophage activation and proliferation depending upon cell type, tissue, disease stage, and context.
During the dysregulated wound healing process, intestinal macrophage not only recruits surrounding mesenchymal cells, such as subepithelial myofibroblasts, to come into the inflamed area but also activates itself and subepithelial myofibroblasts [33,34,35]. Once activated, these cells release a variety of inflammatory cytokines and overproduce extracellular matrix proteins. Finally, these events thicken the tissue layer, destroy the regular motility function and the capability of nutrition absorption in the gut [33,34,35].
Macrophages are essential immune cells for the maintenance of tissue homeostasis in the intestinal mucosa barrier. They are actively involved in the repairing process of wound healing, particularly in the context of intestinal damage and tissue repair in IBD [36]. Phenotypic plasticity of macrophages from classical M1 to alternative M2 is controlled by a variety of cytokines, such as IFN-γ and IL-4. The IL-4-derived M2 can further differentiate into activated myofibroblasts [36]. Alpha-smooth muscle actin (α-SMA) positive-myofibroblasts are central to the wound healing process and highly expressed in patients with fibrostenotic CD [37,38]. They contribute to fibrosis by producing excessive amounts of ECM proteins [33,34].
Our recent study showed that the CD38+/M1 MΦ decreased and CD163+/M2 MΦ increased significantly in macrophages, which were isolated from the colon of 2, 4, 6-Trinitrobenzenesulfonic acid (TNBS)-treated mice compared to ethanol-treated mice [39]. The M2 MΦ was increased in the colon of TNBS treated mice due to MΦ-to-myofibroblast transition, where M1 MΦ decreased significantly. Treatment with tunicamycin significantly increased the ER stress marker, GRP78, and CD163+/M2 MΦ population. Treatment with IL-4 had a similar effect on the numbers of CD163+/M2 MΦ. Treatment with a green tea compound, epigallocatechin-3-gallate (EGCG), an ER stress inhibitor, suppressed IL-4-induced increase in CD163+/M2 MΦ. The effect was blocked with a neutralizing antibody against the 67-kDa laminin receptor (67LR), a reported EGCG-binding receptor. The inhibitory effect of EGCG was associated with an increase in 67LR+/vimentin+ macrophages isolated from mice with TNBS-induced colitis compared to the ethanol-treated group. EGCG also suppressed the tunicamycin-induced increase in GRP78 and production of α-SMA+ macrophages during MΦ-to-myofibroblast transition through its binding to 67LR [39]. These data suggest that ER stress may regulate the phenotypic change of macrophages and macrophage-to-myofibroblast transition. However, the exact role of macrophages during the development of intestinal fibrosis in patients with CD still awaits further study.
5. Epigenetic Regulation of the UPR
The rapidly developing field of epigenetics demonstrates the great potential to elucidate the pathological mechanism of abnormal gene expression due to the changes in the structure and function of the chromatin. These changes can be caused by environmental factors, such as hypoxia, microbial toxins (e.g., Shiga toxigenic factors that degrade GRP78), and dietary factors (e.g., iron) [40,41].
Epigenetic mechanisms affect gene expression and cellular function through three distinctive but also interconnected mechanisms: (1) Chromatin structure modulation, (2) DNA methylation, and (3) RNA interference by small non-coding RNAs, i.e., microRNAs [42,43,44,45].
Llinàs-Arias et al. [46] showed that the small p97/VCP-interacting protein (SVIP), an endogenous inhibitor of ERAD, underwent DNA hypermethylation-associated silencing in high-risk patients who manifest poor clinical outcomes. The dependence of SVIP-hypermethylated cancer cells on aerobic glycolysis and glucose was also related to the sensitivity to an inhibitor of the glucose transporter GLUT1. This study demonstrated that how epigenetics affects ER stress and how SVIP epigenetic silencing in cancer may be applicable to the therapy that targets glucose transporters. Little is known about GRP78 proteostasis and the role of its posttranslational modifications in ER stress. Sieber et al. [47] reported a novel proteostatic mechanism that is dependent on the posttranslational modification of GRP78, allowing cells to differentially regulate protein production during ER stress. ER stress led to de novo biosynthesis of non-trimethylated GRP78, whereas homeostatic, N-lysine methyltransferase 21A (METTL21A)-dependent lysine 585–trimethylated GRP78 was reduced. In other words, ER stress triggered the de novo synthesis of non-trimethylated GRP78 and simultaneous degradation of existing, lysine-trimethylated GRP78. This previously unrecognized mechanism suggests the lack of posttranslational modification may alter the conformation of GRP78 in a way that may be beneficial during ER stress to secure cell survival.
The emergence of miRNAs during the course of UPR-mediated adaptive and apoptotic signaling has provided more mechanistic understanding of their roles in gene regulation in vivo. For example, miR-379 targets (and therefore represses) Edem3, which encodes an inhibitor of ER stress, whereas miR-494, another miRNA in the miR-379 cluster, targets Atf3, a repressor of CHOP [48].
Differential microRNAs’ activities contribute to pro-adaptive/survival and pro-apoptotic UPR signaling by targeting the three main sensors of ER stress, including IRE1, PERK, and ATF6, in vitro and in vivo [49,50]. For example, Upton et al. [51] reported that IRE1α RNase activation caused selective microRNAs (miRs -17, -34a, -96, and -125b) degradation that normally represses translation of Caspase-2 mRNA, leading to activation of the mitochondrial apoptotic pathway.
Moreover, our recent study showed that the UPR and its downstream signaling pathways could be manipulated through epigenetic regulations [9]. We showed that expression of ER stress sensors increased significantly in subepithelial myofibroblasts of strictured intestine from patients with fibrostenotic CD [9]. The increase in the ER stress response featured with overexpression of GRP78, XBP1s, and ATF6α can be reproduced in the normal subepithelial myofibroblasts when treated with tunicamycin, which is an ER stress agonist [9]. The increased levels of ER stress in affected ileum was associated with silencing of miR-199a-5p by DNA-methyltransferase 1 (DNMT1)-mediated promoter hypermethylation [9]. At rest condition, miR-199a-5p targeted ER stressors including GRP78, ATF6, and XBP1s for their degradation [9]. Restoration of miR-199a-5p through a DNA methylation inhibitor, 5-azacytidine, via inhibition of DNMT1 function, suppressed ER stress-induced myofibroblasts activation and excess ECM production [9]. During ER stress, DNMT1 upregulated and led to hypermethylation of miR-199a-5p and its silencing. This silencing in miR-199a-5p led to the loss of its inhibition on the ER components and causes upregulation of the ER stress components, TGF-β1 levels, and resultant fibrosis [9]. Put together, this epigenetic evidence will improve our understanding of the molecular mechanism of fibrosis within the context of ER stress and the UPR (Figure 2).
In summary, epigenetic regulation of ER stress and the UPR may provide a deeper understanding of how a variety of UPR branches and downstream signaling pathways contribute to the pathogenesis of different diseases, suggesting novel pharmacological targets of the ER stress components.
6. Crosstalk between the UPR, Senescence, and Autophagy
6.1. Senescence and ER Stress
Senescence is a cellular state featured with a permanent cell-cycle arrest and molecular changes, including epigenetic, metabolic, membrane lipid composition, and substantial morphological alterations with cell enlargement [52,53,54]. Compared to proliferating cells, senescent cells are not responsive to mitotic stimuli or to apoptosis signals [53,54]. Senescent cells secrete different cytokines, chemokines, growth factors, and matrix remodeling proteases, forming the senescence-associated secretory phenotype (SASP) [53]. SASP can activate immune responses that can either prevent or promote disease development, depending upon specific pathophysiological context [52,53,54]. Cells undergoing senescence upon various types of stress can also promote the UPR activation [53,54]. All three ER sensors, including PERK, IRE1, and ATF6α, activate corresponding downstream signaling events to attenuate protein synthesis as well as induce transcriptional activation. Some of the UPR molecular components activate senescence hallmarks, including cell cycle arrest, DNA repair, morphological change, metabolic alteration, secretory pathway activation, and composition changes in membrane lipids [49,50].
Previous studies suggest that the UPR is associated with senescence at certain levels [53,54]. It should be interesting to better characterize the role of the UPR in the formation of SASP within cell type- and tissue-dependent context, although there are some controversies about that whether the UPR is a consequence of cell senescence or a driver of cell senescence. Interestingly, the gut microbiota is also reported to have its influence on senescence during tumor development in various organs, such as gut, liver, and stomach [52]. However, the role of senescence in the pathogenesis of IBD has not been reported yet.
6.2. Autophagy and ER Stress
The crosstalk between ER stress and autophagy in the pathogenesis of IBD has received a significant amount of attention in recent years [25,55,56]. The UPR and autophagy are interconnected signaling pathways that can compensate for the loss of each other in the intestinal epithelium [55,56].
Adolph et al. [57] showed that Xbp1∆IEC mice demonstrated autophagosome formation in hypomorphic Paneth cells, which is associated with an increase in ER stress response via PERK, eIF2α, and ATF4 pathway to promote autophagy. Moreover, in Atg16l1∆IEC mice with deficient autophagy in the intestinal epithelium, intestinal epithelial apoptosis, IRE1α-regulated NF-κB activation, and TNF signaling were synergistically enhanced [57]. ER stress, autophagy, and spontaneous ileitis take place from Paneth cell-specific deletion of Xbp1 mice [57].
Despite increasing expansion in the number of genetic loci linked to IBD by GWAS, NOD2 (followed by IL23R and ATG16L1) showed a certain fraction of genetic heritability [56]. Autophagy, NOD-like receptor (NLR), and the UPR are functionally interconnected within intestinal epithelia that shares the common dysfunction, which converges upon Paneth cells and myeloid cells, due to deficient ATG16L1, NOD2, and XBP1 activity in transgenic mice and patients with CD-associated NOD255 and ATG16L1 variants [56]. It is also important to note that luminal bacteria have a direct impact on the human epigenome. However, the correlation between this important factor, i.e., NOD and CD phenotype, is still not clear. These findings suggest the crosstalk between the UPR and autophagy exists in intestinal epithelium to maintain intestinal homeostasis.
However, in cancer cells, ER stress-activated autophagy can alleviate UPR and reduce cell death compared to non-transformed cells, which suggests autophagy plays a different role in cell type-dependent manner [58]. Lopes et al. [59] showed that ATF6 enhanced autophagic killing of bacteria, thereby, preventing damage of the epithelial barrier that was caused by dysfunctional mitochondria. Promotion of autophagy amid ER stress seems to protect against further intestinal damage. GWAS identified genetic loci that affect the UPR include those associated with XBP1, AGR2, and ORMDL3, whereas those that affect autophagy include ATG16L1, IRGM, and Leucine-rich repeat kinase 2 (LRRK2). This evidence suggests a link between autophagy and the UPR in the pathogenesis of IBD.
7. The UPR as a Therapeutic Target
As a key player in the immune response during the inflammatory process, the UPR has been investigated as a promising pharmacological target in many different diseases, providing the patients with more optimal choices for personalized medicine [4,5]. Chemical chaperones are considered as low molecular compounds to improve ER protein folding by reducing protein overload. For example, TUDCA and 4-phenyl butyrate (PBA) have been tested in studies or clinical trials for the treatment of different diseases [3,4,5,60].
ER stress inhibitors that promote adaptive UPR signaling and/or prevent the ER stress-mediated cell apoptosis offer another promising therapy target. For example, CHOP inhibitor reduces inflammation-induced lung epithelial cell damage [32]. However, when CHOP inhibitor is applied to mesenchymal cells, it may exacerbate fibrogenesis via activation of myofibroblasts by TGF-β secreted from activated macrophages. Therefore, the cell type-dependent effect of specific ER stress inhibitors should be evaluated to avoid off-target side effects. In addition, proteasome inhibitors, such as bortezomib and MG132, are reported to treat multiple myeloma via blocking the 26S proteasome to stimulate adaptive UPR [3,5].
In 2006, Brownlie et al. [61] reported that the prophylactic or therapeutic parenteral delivery of GRP78/BiP prevented induction of collagen-induced arthritis (CIA) in mice. In 2016, the first human clinical trial using intravenous GRP78/BiP demonstrated that GRP78/ BiP (⩽15 mg) is safe in patients with active rheumatoid arthritis [62]. Patients received a single IV infusion over 1 h and were observed as inpatients overnight. A 12-week follow-up for clinical, rheumatological, and laboratory assessments for safety, efficacy (DAS28-ESR) and biomarker analysis was performed. Good DAS28-ESR responses were achieved in all treatment groups [62]. In phase I/IIA RAGULA trial, 42 patients with rheumatoid arthritis were screened, and 24 were randomized to receive either BiP or placebo. The results showed that after a single IV infusion, BiP might induce remission lasting up to three months in those patients [62].
Given the limited availability of mechanism-based therapies for CD, neutralization of ER stress response and maintenance of the basal UPR using pharmacological molecules represent a promising therapeutic approach towards controlling inflammation and preventing the progression of intestinal fibrosis in those susceptible patients with CD.
8. Future Directions
The ER is a multifunctional signaling organelle that controls a wide range of cellular activities related to the life and death of every single cell under ER stress. The UPR has now been recognized for its important role in regulating inflammatory and immune responses, in cellular and tissue homeostasis, and in immune cell differentiation and function. However, the mechanisms underlying the cell survival to apoptosis transition during ER stress events remain largely unknown. Below, several outstanding questions are listed in Box 1 and await future explorations. Furthermore, mechanistic studies are necessary to elucidate the molecular and cellular mechanisms between senescence and UPR, since data from in vivo models are currently scarce.
Box 1. Outstanding questions.
- What is the direct cause of ER stress or unfolded protein response in human disease?
- How do the different binding partners and modifiers of the UPR components regulate their activity and contribute to cell type- and tissue-specific functions?
- How do cells decide when to initiate apoptosis, at what point, and are these mechanisms important in developmental regulation?
- What is the role of the UPR in adipose tissue and mesenchymal cells where ER stress is less well characterized?
- How do different cytokines affect ER stress response, such as IL-6 and IL-10, for exam-ple?
- Is there a cytokine or any other unknown stimulant that can directly activate ER stress?
- How does misfolded protein in the ER cause oxidative stress?
- How does the UPR establish the crosstalk with senescence and autophagy?
- How do we decide which animal model of ER stress can closely recapitulate the patho-genesis of the disease we study?
9. Conclusions
The UPR is a conserved signaling network that is discovered from yeast to mammalian systems. The UPR is activated in both acute and chronic ER stress with corresponding cellular adaption. Apoptosis is activated to clean the damaged cells when they fail to maintain intracellular homeostasis. As illustrated in Figure 2 of this review, ER stress plays a dual role by inducing apoptosis in intestinal epithelial cells on the one side, and promoting exaggerated adaptive, survival-associated UPR signaling in mesenchymal cells on the other side (Figure 2). Restoration of the ER homeostasis is essential for the treatment of intestinal fibrosis as well as other fibrotic diseases. However, concern should be raised to evaluate the potential pitfall of whether systemic suppression of ER stress is beneficial for patients with a specific phenotype, for example, inflammatory vs. fibrostenotic (overactive wound healing). Selective inhibition of ER stress in a specific cell type, such as mesenchymal cells, to prevent cell proliferation, and in epithelial cells to skip apoptosis-induced mucosal damage might lead to tailored individual therapy. With the development of several therapeutic agents that enhance proteostasis by targeting specific UPR components, the gap between the understanding of the role of UPR and its therapeutic application in patients with immune-mediated diseases will be improved in the near future.
Funding
This research was funded by grants from the Virginia Commonwealth University (VCU)’s CTSA (UL1TR000058 from the National Center for Advancing Translational Sciences) & the CCTR Endowment Fund of VCU and from the Crohn’s & Colitis Foundation (Ref. #550514).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The author declares no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| α-SMA | alpha-smooth muscle actin |
| ATF | activating transcription factor |
| BiP | immunoglobulin heavy chain-binding protein |
| CHOP | C⁄EBP homologous protein |
| DNMT1 | DNA-methyltransferase 1 |
| eIF2 a | a-subunit of eukaryotic translational initiation factor 2 |
| EGCG | epigallocatechin-3-gallate |
| ER | endoplasmic reticulum |
| ERAD | ER-associated degradation; |
| GRP78 | glucose-regulated protein 78 kDa |
| GWAS | Genome-wide association studies |
| IRE1 | inositol requirement 1 |
| JNK | Jun N-terminal kinase |
| IBD | Inflammatory Bowel Disease |
| LRRK2 | Leucine-rich repeat kinase 2; |
| NLR | NOD-like receptor |
| ORMDL3 | Orosmucoid-like 3 |
| PBA | 4-phenyl butyrate |
| PDI | protein disulfide isomerase |
| PERK | PRKR-like endoplasmic reticulum kinase |
| PKR | double stranded RNA-dependent protein kinase |
| SASP | senescence-associated secretory phenotype |
| SVIP | small p97/VCP-interacting protein |
| TNF | tumor necrosis factor |
| TUDCA | tauroursodeoxycholic acid |
| XBP1 | x-box binding protein 1 |
References
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug. Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Dai, Z.; Sun, K.; Zhang, Y.; Chen, J.; Yang, Y.; Tso, P.; Wu, G.; Wu, Z. Intestinal Epithelial Cell Endoplasmic Reticulum Stress and Inflammatory Bowel Disease Pathogenesis: An Update Review. Front. Immunol. 2017, 8, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138. [Google Scholar] [CrossRef]
- Li, C.; Grider, J.R.; Murthy, K.S.; Bohl, J.; Rivet, E.; Wieghard, N.; Kuemmerle, J.F. Endoplasmic Reticulum Stress in Subepithelial Myofibroblasts Increases the TGF-β1 Activity That Regulates Fibrosis in Crohn’s Disease. Inflamm. Bowel Dis. 2020, 26, 809–819. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Investig. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulsky, A.V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 2017, 13, 681–696. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, D.P.; Gardet, A.; Törkvist, L.; Goyette, P.; Essers, J.; Taylor, K.D.; Neale, B.M.; Ong, R.T.H.; Lagacé, C.; Li, C.; et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010, 42, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Kaplan GG.Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Roda, G.; Chien, N.S.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Primers 2020, 6, 22. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Moffatt, M.F.; Kabesch, M.; Liang, L.; Dixon, A.L.; Strachan, D.; Heath, S.; Depner, M.; von Berg, A.; Bufe, A.; Rietschel, E.; et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007, 448, 470–473. [Google Scholar] [CrossRef]
- Pranculienė, G.; Steponaitienė, R.; Skiecevičienė, J.; Kučinskienė, R.; Kiudelis, G.; Adamonis, K.; Labanauskas, L.; Kupčinskas, L. Associations between NOD2, IRGM and ORMDL3 polymorphisms and pediatric-onset inflammatory bowel disease in the Lithuanian population. Medicina (Kaunas) 2016, 52, 325–330. [Google Scholar] [CrossRef]
- Zheng, W.; Rosenstiel, P.; Huse, K.; Sina, C.; Valentonyte, R.; Mah, N.; Zeitlmann, L.; Grosse, J.; Ruf, N.; Hampe, J. Evaluation of AGR2 and AGR3 as candidate genes for inflammatory bowel disease. Genes Immun. 2006, 7, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Maurel, M.; Obacz, J.; Avril, T.; Ding, Y.P.; Papadodima, O.; Treton, X.; Daniel, F.; Pilalis, E.; Hörberg, J.; Ogier-Denis, E.; et al. Control of anterior GRadient 2 (AGR2) dimerization links endoplasmic reticulum proteostasis to inflammation. EMBO Mol. Med. 2019, 11, e10120. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Blumberg, R.S.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Shkoda, A.; Ruiz, P.A.; Daniel, H.; Kim, S.C.; Rogler, G.; Sartor, R.B.; Haller, D. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: Impact on chronic inflammation. Gastroenterology 2007, 132, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Hooper, K.M.; Barlow, P.G.; Henderson, P.; Stevens, C. Interactions between Autophagy and the Unfolded Protein Response: Implications for Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 661–671. [Google Scholar] [CrossRef]
- Cao, S.S.; Wang, M.; Harrington, J.C.; Chuang, B.M.; Eckmann, L.; Kaufman, R.J. Phosphorylation of eIF2α is dispensable for differentiation but required at a posttranscriptional level for paneth cell function and intestinal homeostasis in mice. Inflamm. Bowel Dis. 2014, 20, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Zimmermann, E.M.; Chuang, B.M.; Song, B.; Nwokoye, A.; Wilkinson, J.E.; Eaton, K.A.; Kaufman, R.J. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 2013, 144, 989–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Ishitsuka, Y.; Hayasaka, M.; Yamada, Y.; Miyata, K.; Endo, M.; Kondo, Y.; Moriuchi, H.; Irikura, M.; Tanaka, K.; et al. The exacerbating roles of CCAAT/enhancer-binding protein homologous protein (CHOP) in the development of bleomycin-induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice. Pharmacol. Res. 2015, 99, 52–62. [Google Scholar] [CrossRef]
- Endo, M.; Mori, M.; Akira, S.; Gotoh, T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J. Immunol. 2006, 176, 6245–6253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayaub, E.A.; Kolb, P.S.; Mohammed-Ali, Z.; Tat, V.; Murphy, J.; Bellaye, P.S.; Shimbori, C.; Boivin, F.J.; Lai, R.; Lynn, E.G.; et al. GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J. Pathol. 2016, 239, 411–425. [Google Scholar] [CrossRef]
- Malhi, H.; Kropp, E.M.; Clavo, V.F.; Kobrossi, C.R.; Han, J.; Mauer, A.S.; Yong, J.; Kaufman, R.J. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J. Biol. Chem. 2013, 288, 18624–18642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Wang, Y.; Zhang, Z.; He, L.; Zhu, J.; Zhang, M.; He, X.; Cheng, Z.; Ao, Q.; Cao, Y.; et al. Chop Deficiency Protects Mice Against Bleomycin-induced Pulmonary Fibrosis by Attenuating M2 Macrophage Production. Mol. Ther. 2016, 24, 915–925. [Google Scholar] [CrossRef]
- Bain, C.C.; Mowat, A.M. Macrophages in intestinal homeostasis and inflammation. Immunol. Rev. 2014, 260, 102–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hine, A.M.; Loke, P. Intestinal Macrophages in Resolving Inflammation. J. Immunol. 2019, 203, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Tang, P.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kuemmerle, J.F. The fate of myofibroblasts during the development of fibrosis in Crohn’s disease. J. Dig. Dis. 2020, 21, 326–331. [Google Scholar] [CrossRef]
- Mifflin, R.C.; Pinchuk, I.V.; Saada, J.I.; Powell, D.W. Intestinal myofibroblasts: Targets for stem cell therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G684–G696. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Kuemmerle, J.F. Endoplasmic reticulum stress-mediated macrophage-to-myofibroblast transition contributes to intestinal fibrosis in murine TNBS-induced colitis. In Gastroenterology; W.B. Saunders Co-Elsevier: Philadelphia, PA, USA, 2020. [Google Scholar]
- Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxins: Intracellular trafficking to the ER leading to activation of host cell stress responses. Toxins 2010, 2, 1515–1535. [Google Scholar] [CrossRef] [Green Version]
- Werner, T.; Wagner, S.J.; Martinez, I.; Walter, J.; Chang, J.; Clavel, T.; Kisling, S.; Schuemann, K.; Haller, D. Depletion of luminal iron alters the gut microbiota and prevents Crohn’s disease-like ileitis. Gut 2011, 60, 325–333. [Google Scholar] [CrossRef]
- Li, C.; Kuemmerle, J. Epigenetic regulation of intestinal fibrosis. In Firbrostenotic Inflammatory Bowel Disease, 1st ed.; Rieder, F., Ed.; Springer: Cham, Switzerland, 2018; Chapter 4; pp. 39–58. [Google Scholar]
- Li, C.; Kuemmerle, J.F. Genetic and epigenetic regulation of intestinal fibrosis. United Eur. Gastroenterol. J. 2016, 4, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.; Mann, D.A. Epigenetic regulation of wound healing and fibrosis. Curr. Opin. Rheumatol. 2013, 25, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Natarajan, R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat. Rev. Nephrol. 2019, 15, 327–345. [Google Scholar] [CrossRef]
- Llinàs-Arias, P.; Rosselló-Tortella, M.; López-Serra, P.; Pérez-Salvia, M.; Setién, F.; Marin, S.; Muñoz, J.P.; Junza, A.; Capellades, J.; Esteller, M.; et al. Epigenetic loss of the endoplasmic reticulum-associated degradation inhibitor SVIP induces cancer cell metabolic reprogramming. JCI Insight 2019, 5, e125888. [Google Scholar] [CrossRef] [Green Version]
- Sieber, J.; Wieder, N.; Ostrosky-Frid, M.; Dvela-Levitt, M.; Aygün, O.; Udeshi, N.D.; Carr, S.A.; Greka, A. Lysine trimethylation regulates 78-kDa glucose-regulated protein proteostasis during endoplasmic reticulum stress. J. Biol. Chem. 2017, 292, 18878–18885. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Wang, M.; Chen, Z.; Bhatt, K.; Oh, H.J.; Lanting, L.; Deshpande, S.; Jia, Y.; Lai, J.Y.; Natarajan, R.; et al. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat. Commun. 2016, 7, 12864. [Google Scholar] [CrossRef]
- Byrd, A.E.; Brewer, J.W. Micro(RNA)managing endoplasmic reticulum stress. IUBMB Life 2013, 65, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C. Intestinal fibrosis in inflammatory bowel disease: The role of microRNAs. Explor. Res. Hypothese Med. 2015, 1, 7–16. [Google Scholar]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Oakes, S.A.; et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Frey, N.; Venturelli, S.; Zender, L.; Bitzer, M. Cellular senescence in gastrointestinal diseases: From pathogenesis to therapeutics. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [Green Version]
- Abbadie, C.; Pluquet, O. Unfolded Protein Response (UPR) Controls Major Senescence Hallmarks. Trends Biochem. Sci. 2020, 45, 371–374. [Google Scholar] [CrossRef]
- Hosomi, S.; Kaser, A.; Blumberg, R.S. Role of endoplasmic reticulum stress and autophagy as interlinking pathways in the pathogenesis of inflammatory bowel disease. Curr. Opin. Gastroenterol. 2015, 31, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaser, A.; Blumberg, R.S. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology 2011, 140, 1738–1747. [Google Scholar] [CrossRef] [Green Version]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [Green Version]
- Lopes, F.; Keita, Å.V.; Saxena, A.; Reyes, J.L.; Mancini, N.L.; Al Rajabi, A.; Wang, A.; Baggio, C.H.; Dicay, M.; McKay, D.M.; et al. ER-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J. Biol. Chem. 2018, 293, 3073–3087. [Google Scholar] [CrossRef] [Green Version]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling-from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Brownlie, R.J.; Myers, L.K.; Wooley, P.H.; Corrigall, V.M.; Bodman-Smith, M.D.; Panayi, G.S.; Thompson, S.J. Treatment of murine collagen-induced arthritis by the stress protein BiP via interleukin-4-producing regulatory T cells: A novel function for an ancient protein. Arthritis Rheum. 2006, 54, 854–863. [Google Scholar] [CrossRef]
- Kirkham, B.; Chaabo, K.; Hall, C.; Garrood, T.; Mant, T.; Allen, E.; Vincent, A.; Vasconcelos, J.C.; Prevost, A.T.; Panayi, G.S.; et al. Safety and patient response as indicated by biomarker changes to binding immunoglobulin protein in the phase I/IIA RAGULA clinical trial in rheumatoid arthritis. Rheumatology 2016, 55, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
A word cloud of key concepts presented in this review is made by WordItOut online software. https://worditout.com/word-cloud/create.
Figure 1.
A word cloud of key concepts presented in this review is made by WordItOut online software. https://worditout.com/word-cloud/create.
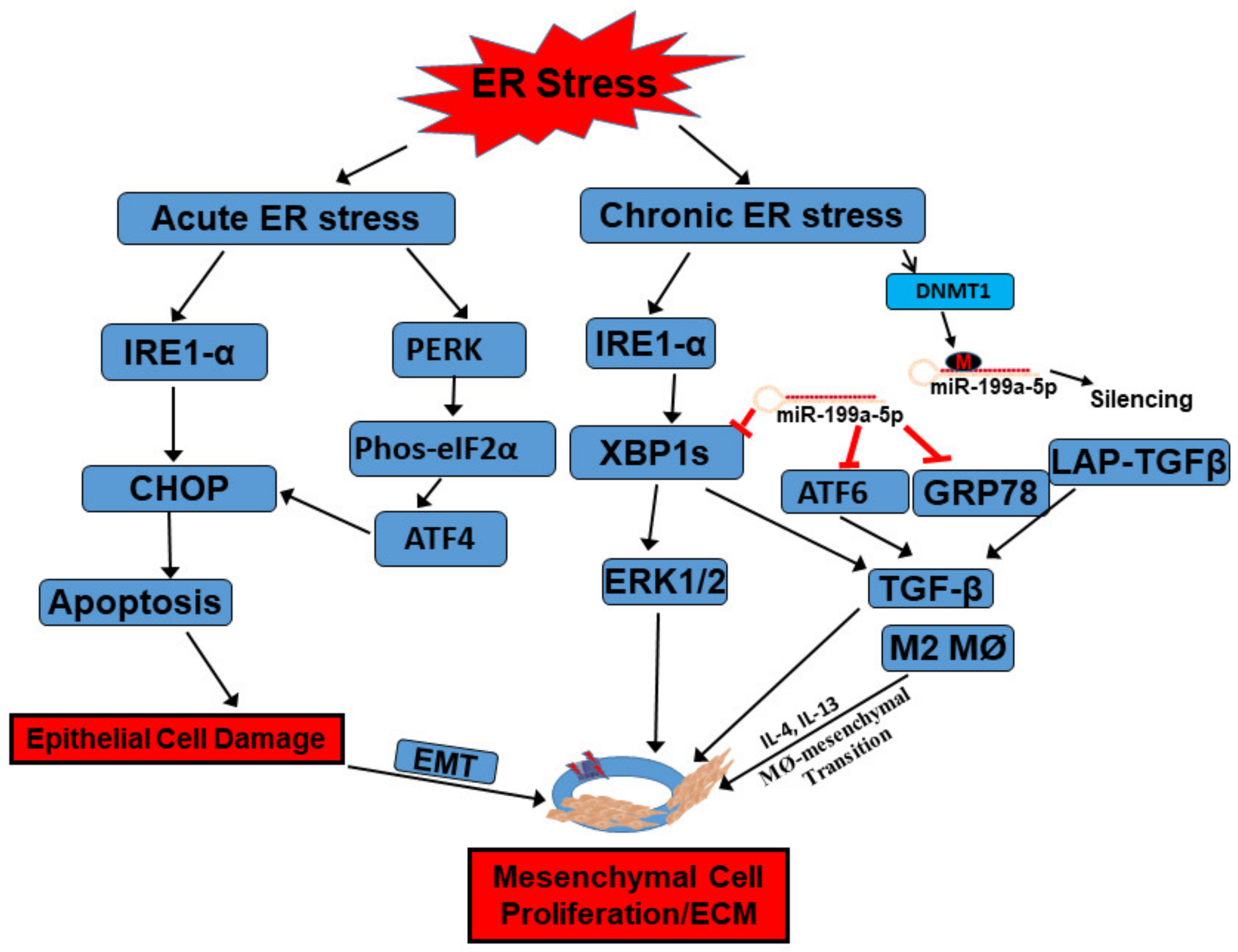
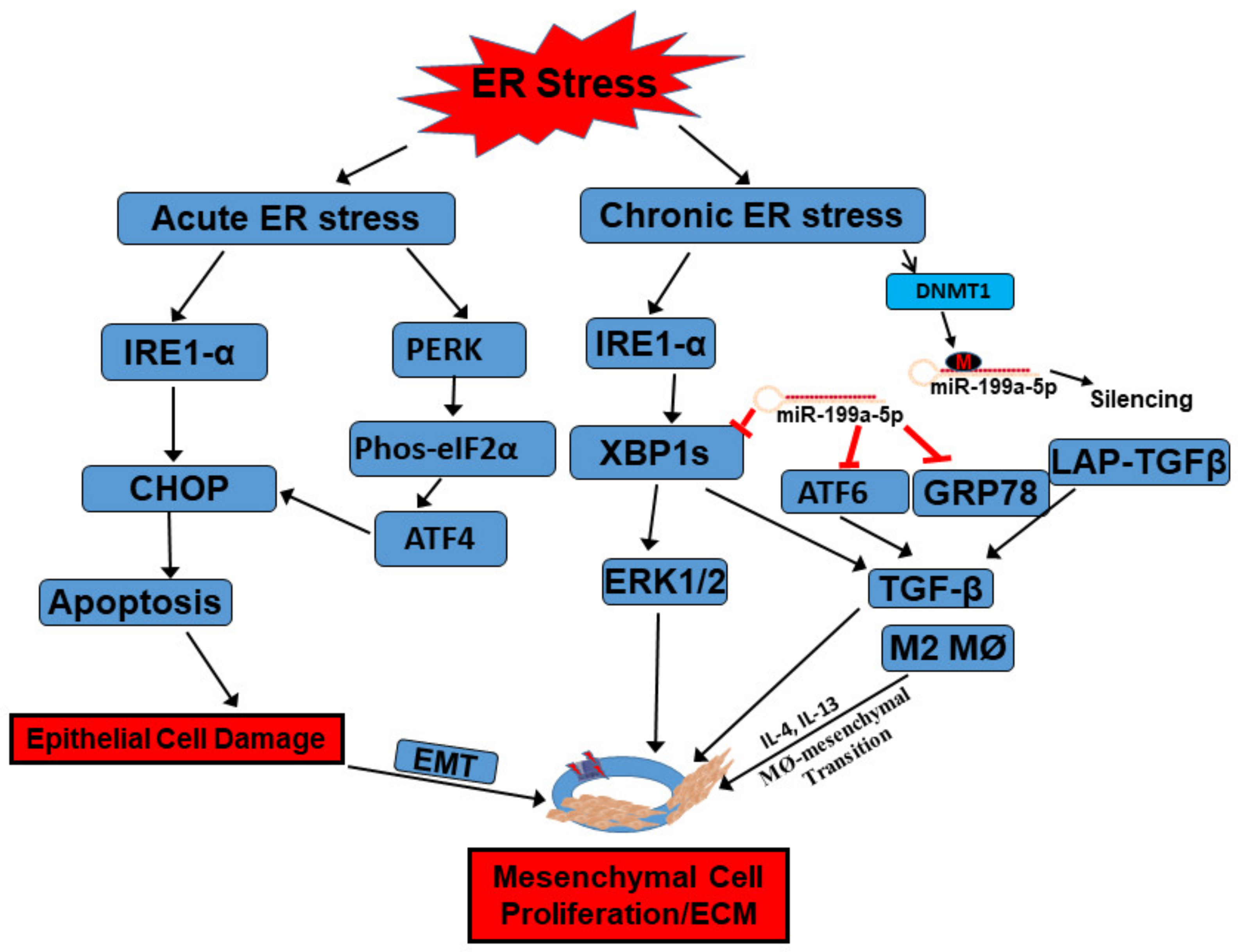
Figure 2.
Functions of the UPR in the development of intestinal fibrosis in Crohn’s disease. When intestinal epithelial cells (IECs) are subject to acute ER stress, ER stress sensors IRE1α and PERK can be activated with detached association from binding partner GRP78. Downstream signaling, including CHOP and eIF2α/ATF4, are further activated to induce apoptosis in IECs. Meanwhile, inflammatory cytokines, such as IL-4 and IL-13, induced activation of macrophages (M2) as well as macrophage-to-mesenchymal transition. TGF-β can be secreted from this transition to further activate mesenchymal cells, such as subepithelial myofibroblasts, to proliferate and induce extracellular matrix protein production. When the intestine is subject to chronic inflammation-induced ER stress, IRE1α catalyzes non-canonical splicing of X-box binding protein 1 (XBP1) mRNA into the constitutively active form XBP1s, which activates ERK1/2 to stimulate mesenchymal cell proliferation. The increased UPR is also associated with increased silencing of miR-199a-5p by DNMT1-mediated promoter hypermethylation. At rest condition, miR-199a-5p targets different ER stressors, including GRP78, ATF6, and XBP1s, through complementary binding to their promoter regions for their degradation. ATF6 and XBP1 both serve as transcription factors and activate ER stress-induced myofibroblasts activation through upregulation of TGF-β and excess ECM production. GRP78 can bind to latent associated peptide (LAP)-TGFβ to activate TGF-β. All these factors can finally contribute to the development of intestinal fibrosis. Refer to context for details.
Figure 2.
Functions of the UPR in the development of intestinal fibrosis in Crohn’s disease. When intestinal epithelial cells (IECs) are subject to acute ER stress, ER stress sensors IRE1α and PERK can be activated with detached association from binding partner GRP78. Downstream signaling, including CHOP and eIF2α/ATF4, are further activated to induce apoptosis in IECs. Meanwhile, inflammatory cytokines, such as IL-4 and IL-13, induced activation of macrophages (M2) as well as macrophage-to-mesenchymal transition. TGF-β can be secreted from this transition to further activate mesenchymal cells, such as subepithelial myofibroblasts, to proliferate and induce extracellular matrix protein production. When the intestine is subject to chronic inflammation-induced ER stress, IRE1α catalyzes non-canonical splicing of X-box binding protein 1 (XBP1) mRNA into the constitutively active form XBP1s, which activates ERK1/2 to stimulate mesenchymal cell proliferation. The increased UPR is also associated with increased silencing of miR-199a-5p by DNMT1-mediated promoter hypermethylation. At rest condition, miR-199a-5p targets different ER stressors, including GRP78, ATF6, and XBP1s, through complementary binding to their promoter regions for their degradation. ATF6 and XBP1 both serve as transcription factors and activate ER stress-induced myofibroblasts activation through upregulation of TGF-β and excess ECM production. GRP78 can bind to latent associated peptide (LAP)-TGFβ to activate TGF-β. All these factors can finally contribute to the development of intestinal fibrosis. Refer to context for details.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, C. Unfolded Protein Response and Crohn’s Diseases: A Molecular Mechanism of Wound Healing in the Gut. Gastrointest. Disord. 2021, 3, 31-43. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord3010004
AMA Style
Li C. Unfolded Protein Response and Crohn’s Diseases: A Molecular Mechanism of Wound Healing in the Gut. Gastrointestinal Disorders. 2021; 3(1):31-43. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord3010004
Chicago/Turabian StyleLi, Chao. 2021. "Unfolded Protein Response and Crohn’s Diseases: A Molecular Mechanism of Wound Healing in the Gut" Gastrointestinal Disorders 3, no. 1: 31-43. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord3010004