The HGF/SF Mouse Model of UV-Induced Melanoma as an In Vivo Sensor for Metastasis-Regulating Gene

 and
and 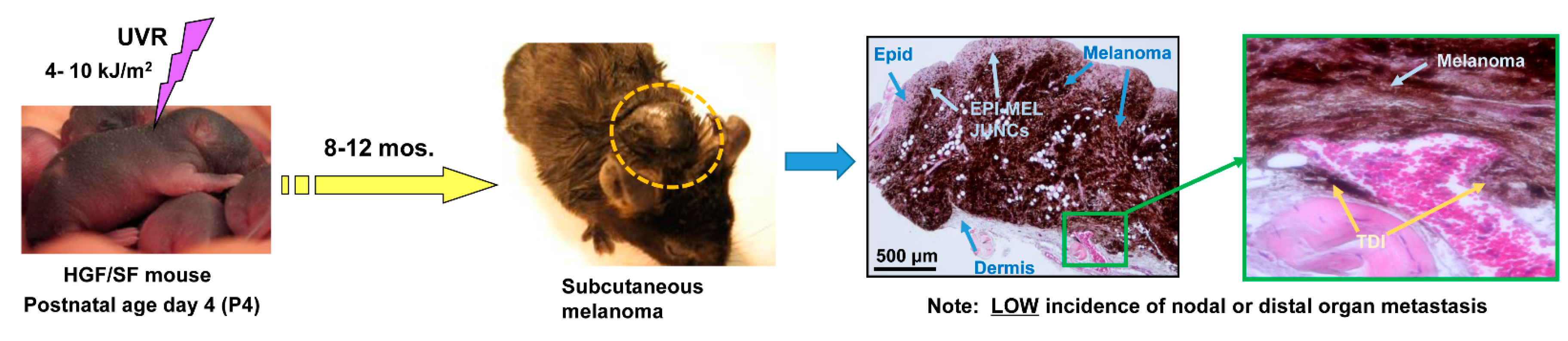{kind=link}
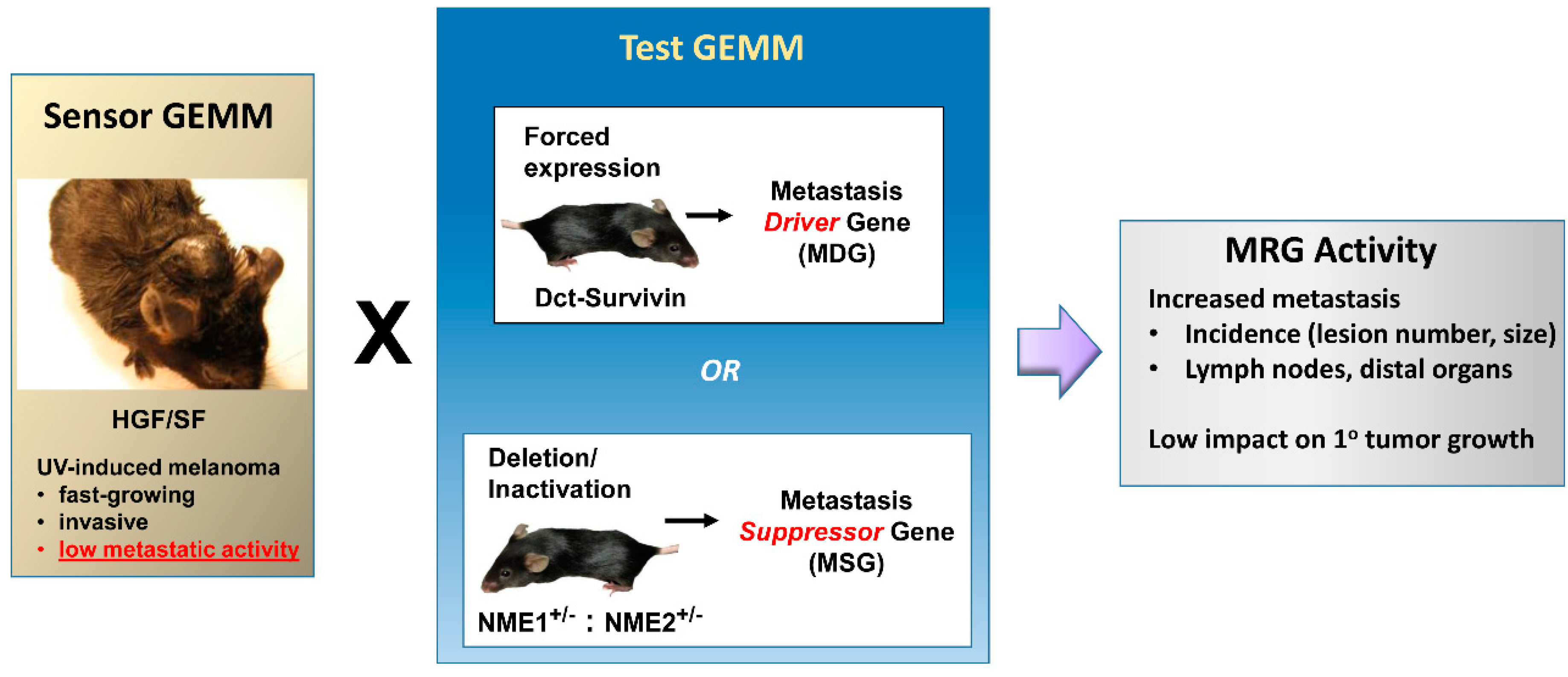{kind=link}
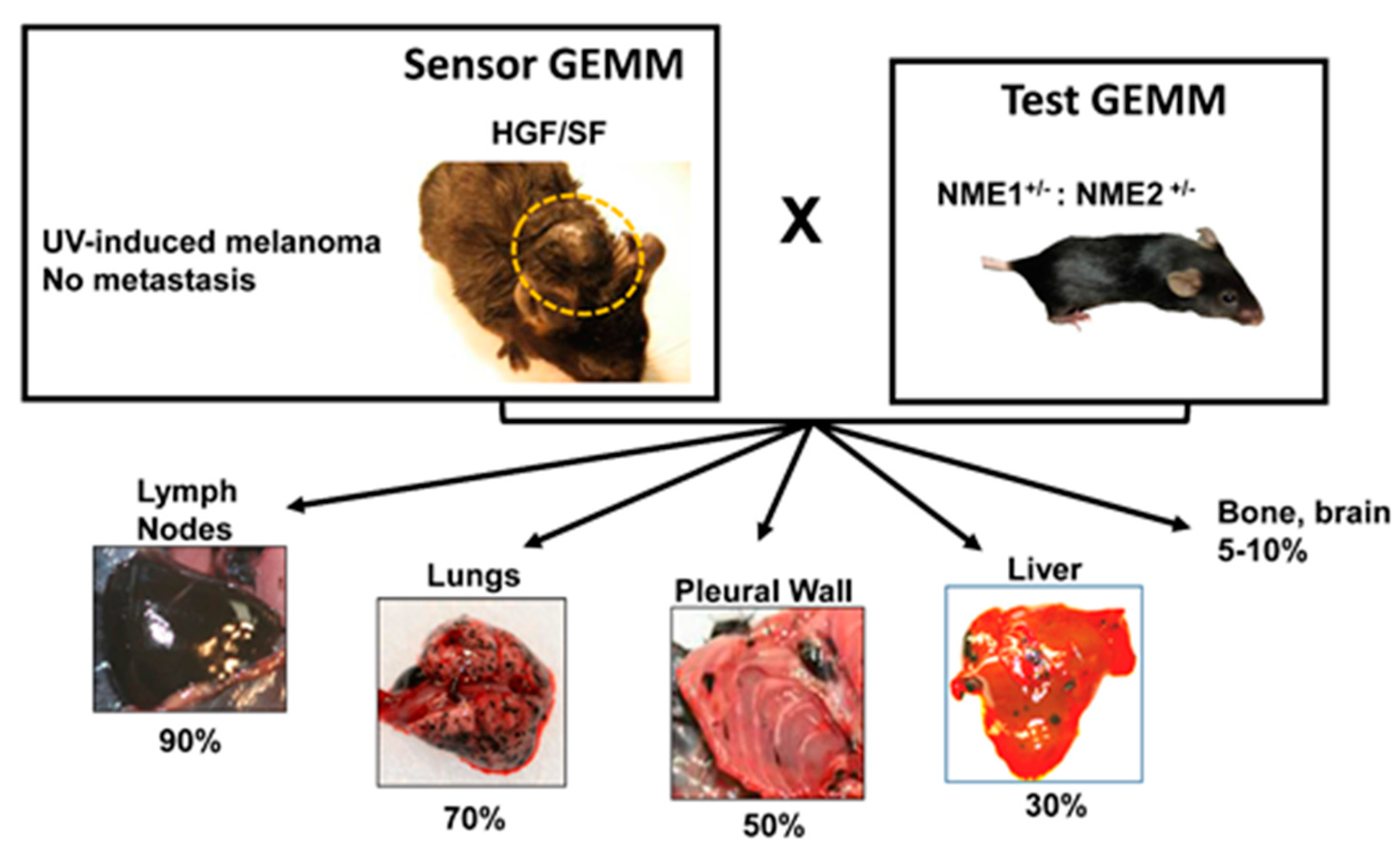{kind=link}
Abstract
:1. Introduction
2. Application of the Hepatocyte Growth Factor/Scatter Factor (HGF/SF) Transgenic Mouse Model as a Sensor of Metastasis-Regulating Genes (MRG) Activity: Working Principles
3. Validation of MRGs Using the HGF/SF Model
3.1. Validation of Two Metastasis Driver Genes (MDGs): CDK4R24C and Survivin
3.1.1. CDK4R24C
3.1.2. Survivin
3.2. MSGs: NME1 and NME2
4. Optimization of the HGF/SF Model as a Sensor for MRG Activity
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CDK | Cyclin-Dependent Kinase |
| Dct | Dopachrome Tautomerase |
| GEMM | Genetically-Engineered Mouse model |
| IAP | Inhibitor of Apoptosis |
| HGF/SF | Hepatocyte Growth Factor/Scatter Growth Factor |
| MRGs | Metastasis-Regulating Genes |
| MDG | Metastasis-Driving Gene |
| MSG | Metastasis Suppressor Gene |
| MT | Metallothionine |
| UVR | Ultraviolet Light Radiation |
| DMBA | 7,12-Dimethylbenz[a]anthracene |
| TPA | 12-O-Tetradecanoylphorbol-13-acetate |
| NF-κB | Nuclear Factor Kappa-light-chain-enhancer of activated B cells |
| LPA | Lysophosphatidic Acid |
| PDGFA | Platelet Derived Growth Factor A |
References
- Shaikh, W.R.; Dusza, S.W.; Weinstock, M.A.; Oliveria, S.A.; Geller, A.C.; Halpern, A.C. Melanoma thickness and survival trends in the United States, 1989–2009. J. Nat. Cancer Inst. 2016, 108, djv294. [Google Scholar] [CrossRef] [PubMed]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; van den Boorn-Konijnenberg, D.; Homig-Holzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Nguyen, F.D.; Noory, M.A.; Sharma, A. Current state of animal (mouse) modeling in melanoma research. Cancer Growth Metastasis 2015, 8, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; LaRochelle, W.J.; Sharp, R.; Otsuka, T.; Kriebel, P.; Anver, M.; Aaronson, S.A.; Merlino, G. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc. Natl. Acad. Sci. USA 1997, 94, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Noonan, F.P.; Recio, J.A.; Takayama, H.; Duray, P.; Anver, M.R.; Rush, W.L.; de Fabo, E.C.; Merlino, G. Neonatal sunburn and melanoma in mice. Nature 2001, 413, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Noonan, F.P.; Zaidi, M.R.; Wolnicka-Glubisz, A.; Anver, M.R.; Bahn, J.; Wielgus, A.; Cadet, J.; Douki, T.; Mouret, S.; Tucker, M.A.; et al. Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment. Nat. Commun. 2012, 3, 884. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, T.; Yoshida, H.; Yamazaki, H.; Miyamoto, A.; Hemmi, H.; Nishimura, E.; Shultz, L.D.; Nishikawa, S.; Hayashi, S. Transgene expression of steel factor in the basal layer of epidermis promotes survival, proliferation, differentiation and migration of melanocyte precursors. Development 1998, 125, 2915–2923. [Google Scholar] [PubMed]
- D’Orazio, J.A.; Nobuhisa, T.; Cui, R.; Arya, M.; Spry, M.; Wakamatsu, K.; Igras, V.; Kunisada, T.; Granter, S.R.; Nishimura, E.K.; et al. Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature 2006, 443, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Vanover, J.C.; Spry, M.L.; Hamilton, L.; Wakamatsu, K.; Ito, S.; D’Orazio, J.A. Stem cell factor rescues tyrosinase expression and pigmentation in discreet anatomic locations in albino mice. Pigment Cell Melanoma Res. 2009, 22, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, T.; Takayama, H.; Sharp, R.; Celli, G.; LaRochelle, W.J.; Bottaro, D.P.; Ellmore, N.; Vieira, W.; Owens, J.W.; Anver, M.; et al. c-Met autocrine activation induces development of malignant melanoma and acquisition of the metastatic phenotype. Cancer Res. 1998, 58, 5157–5167. [Google Scholar] [PubMed]
- Recio, J.A.; Noonan, F.P.; Takayama, H.; Anver, M.R.; Duray, P.; Rush, W.L.; Lindner, G.; de Fabo, E.C.; DePinho, R.A.; Merlino, G. INK4A/ARF deficiency promotes ultraviolet radiation-induced melanomagenesis. Cancer Res. 2002, 62, 6724–6730. [Google Scholar] [PubMed]
- Sharpless, E.; Chin, L. The INK4a/ARF locus and melanoma. Oncogene 2003, 22, 3092–3098. [Google Scholar] [CrossRef] [PubMed]
- Barnhill, R.L.; Lugassy, C. Angiotropic malignant melanoma and extravascular migratory metastasis: Description of 36 cases with emphasis on a new mechanism of tumour spread. Pathology 2004, 36, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Liu, T.; Cotter, M.A.; Florell, S.R.; Robinette, K.; Hanks, A.N.; Grossman, D. Melanocyte expression of survivin promotes development and metastasis of UV-induced melanoma in HGF-transgenic mice. Cancer Res. 2007, 67, 5172–5178. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Novak, M.; Harris, N.; Merlino, G.; Slominski, A.; Kaetzel, D.M. NM23 deficiency promotes metastasis in a UV radiation-induced mouse model of human melanoma. Clin. Exp. Metastasis 2013, 30, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.; Hobbs, C.; Nogueira, C.; Cordon-Cardo, C.; McKee, P.H.; Chin, L.; Bosenberg, M.W. Amplification of CDK4 and MDM2 in malignant melanoma. Genes Chromosomes Cancer 2006, 45, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Wolfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wolfel, C.; Klehmann-Hieb, E.; de Plaen, E.; Hankeln, T.; Meyer zum Buschenfelde, K.H.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Weger, J.; Yang, Q.; Goldstein, A.M.; Tucker, M.A.; Walker, G.J.; Hayward, N.; Dracopoli, N.C. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet. 1996, 12, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Lukas, J.; Guldberg, P.; Alsner, J.; Kirkin, A.F.; Zeuthen, J.; Bartek, J. The p16-cyclin D/Cdk4-pRb pathway as a functional unit frequently altered in melanoma pathogenesis. Cancer Res. 1996, 56, 5475–5483. [Google Scholar] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, R.; Garcia, J.F.; Ortega, S.; Martin, J.; Dubus, P.; Barbacid, M.; Malumbres, M. Invasive melanoma in Cdk4-targeted mice. Proc. Natl. Acad. Sci. USA 2001, 98, 13312–13317. [Google Scholar] [CrossRef] [PubMed]
- Steitz, J.; Buchs, S.; Tormo, D.; Ferrer, A.; Wenzel, J.; Huber, C.; Wolfel, T.; Barbacid, M.; Malumbres, M.; Tuting, T. Evaluation of genetic melanoma vaccines in cdk4-mutant mice provides evidence for immunological tolerance against authochthonous melanomas in the skin. Int. J. Cancer 2006, 118, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N. New developments in melanoma genetics. Curr. Oncol. Rep. 2000, 2, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Begg, C.B.; Orlow, I.; Hummer, A.J.; Armstrong, B.K.; Kricker, A.; Marrett, L.D.; Millikan, R.C.; Gruber, S.B.; Anton-Culver, H.; Zanetti, R.; et al. Lifetime risk of melanoma in CDKN2A mutation carriers in a population-based sample. J. Natl. Cancer Inst. 2005, 97, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Tormo, D.; Ferrer, A.; Gaffal, E.; Wenzel, J.; Basner-Tschakarjan, E.; Steitz, J.; Heukamp, L.C.; Gutgemann, I.; Buettner, R.; Malumbres, M.; et al. Rapid growth of invasive metastatic melanoma in carcinogen-treated hepatocyte growth factor/scatter factor-transgenic mice carrying an oncogenic CDK4 mutation. Am. J. Pathol. 2006, 169, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Gaffal, E.; Landsberg, J.; Bald, T.; Sporleder, A.; Kohlmeyer, J.; Tuting, T. Neonatal UVB exposure accelerates melanoma growth and enhances distant metastases in Hgf-Cdk4R24C C57BL/6 mice. Int. J. Cancer 2011, 129, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Merlino, G. Constitutive c-Met signaling through a nonautocrine mechanism promotes metastasis in a transgenic transplantation model. Cancer Res. 2002, 62, 2951–14956. [Google Scholar] [PubMed]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, E.C.; Baird, S.; Korneluk, R.G.; MacKenzie, A.E. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17, 3247–3259. [Google Scholar] [CrossRef] [PubMed]
- Bowen, A.R.; Hanks, A.N.; Murphy, K.J.; Florell, S.R.; Grossman, D. Proliferation, apoptosis, and survivin expression in keratinocytic neoplasms and hyperplasias. Am. J. Dermatopathol. 2004, 26, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Grossman, D.; McNiff, J.M.; Li, F.; Altieri, D.C. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J. Investig. Dermatol. 1999, 113, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Adida, C.; Berrebi, D.; Peuchmaur, M.; Reyes-Mugica, M.; Altieri, D.C. Anti-apoptosis gene, survivin, and prognosis of neuroblastoma. Lancet 1998, 351, 882–883. [Google Scholar] [CrossRef]
- Lu, C.D.; Altieri, D.C.; Tanigawa, N. Expression of a novel antiapoptosis gene, survivin, correlated with tumor cell apoptosis and p53 accumulation in gastric carcinomas. Cancer Res. 1998, 58, 1808–1812. [Google Scholar] [PubMed]
- Kawasaki, H.; Altieri, D.C.; Lu, C.D.; Toyoda, M.; Tenjo, T.; Tanigawa, N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998, 58, 5071–5074. [Google Scholar] [PubMed]
- Gradilone, A.; Gazzaniga, P.; Ribuffo, D.; Scarpa, S.; Cigna, E.; Vasaturo, F.; Bottoni, U.; Innocenzi, D.; Calvieri, S.; Scuderi, N.; et al. Survivin, bcl-2, bax, and bcl-X gene expression in sentinel lymph nodes from melanoma patients. J. Clin. Oncol. 2003, 21, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Morton, D.L.; Elashoff, D.; Hoon, D.S.B. Survivin expression by metastatic melanoma predicts poor disease outcome in patients receiving adjuvant polyvalent vaccine. Int. J. Cancer 2005, 117, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Vong, Q.P.; Cao, K.; Li, H.Y.; Iglesias, P.A.; Zheng, Y. Chromosome alignment and segregation regulated by ubiquitination of survivin. Science 2005, 310, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Grossman, D.; Kim, P.J.; Schechner, J.S.; Altieri, D.C. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc. Natl. Acad. Sci. USA 2001, 98, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Pennati, M.; Binda, M.; de Cesare, M.; Pratesi, G.; Folini, M.; Citti, L.; Daidone, M.G.; Zunino, F.; Zaffaroni, N. Ribozyme-mediated down-regulation of survivin expression sensitizes human melanoma cells to topotecan in vitro and in vivo. Carcinogenesis 2004, 25, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Hoon, D.B.; Wang, H.C.; Ramshaw, I.A. Increased metastatic ability and bone formation of a mammary adenocarcinoma in vivo after in vitro passaging. Eur. J. Cancer Clin. Oncol. 1984, 20, 1517–1526. [Google Scholar] [CrossRef]
- McKenzie, J.A.; Liu, T.; Jung, J.Y.; Jones, B.B.; Ekiz, H.A.; Welm, A.L.; Grossman, D. Survivin promotion of melanoma metastasis requires upregulation of α 5 integrin. Carcinogenesis 2013, 34, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Mukherjee, N.; Schwan, J.V.; Fujita, M.; Norris, D.A.; Shellman, Y.G. Alternative treatments for melanoma: Targeting BCL-2 family members to de-bulk and kill cancer stem cells. J. Investig. Dermatol. 2015, 135, 2155–2161. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Hu, D.; Tu, L.; Zhou, X.; Lu, F.; Wen, B.; Wu, W.; Lin, Y.; Zhou, Z.; Qu, J. Involvement of PI3K/Akt signaling pathway in hepatocyte growth factor-induced migration of uveal melanoma cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S.; Bevilacqua, G.; Kopper, L.; Thorgerisson, U.R.; Talmadge, J.E.; Liotta, L.A.; Sobel, M. Evidence for a novel gene associated with low tumor metastatic potential. J. Natl. Cancer Inst. 1988, 80, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Hartsough, M.T.; Steeg, P.S. Nm23/nucleoside diphosphate kinase in human cancers. J. Bioenerg. Biomembr. 2000, 32, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Flatow, U.; King, C.R.; Sandeen, M.A.; Margulies, I.M.; Liotta, L.A.; Steeg, P.S. Reduced tumor incidence, metastatic potential, and cytokine responsiveness of nm23-transfected melanoma cells. Cell 1991, 65, 25–35. [Google Scholar] [CrossRef]
- Florenes, V.A.; Aamdal, S.; Myklebost, O.; Maelandsmo, G.M.; Bruland, O.S.; Fodstad, O. Levels of nm23 messenger RNA in metastatic malignant melanomas: Inverse correlation to disease progression. Cancer Res. 1992, 52, 6088–6091. [Google Scholar] [PubMed]
- Smith, S.C.; Theodorescu, D. Learning therapeutic lessons from metastasis suppressor proteins. Nat. Rev. Cancer 2009, 9, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.R.; Welch, D.R. Metastasis suppressor genes at the interface between the environment and tumor cell growth. Int. Rev. Cell Mol. Biol. 2011, 286, 107–180. [Google Scholar] [CrossRef] [PubMed]
- Marino, N.; Nakayama, J.; Collins, J.W.; Steeg, P.S. Insights into the biology and prevention of tumor metastasis provided by the Nm23 metastasis suppressor gene. Cancer Metastasis Rev. 2012, 31, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Boissan, M.; Dabernat, S.; Peuchant, E.; Schlattner, U.; Lascu, I.; Lacombe, M.L. The mammalian Nm23/NDPK family: From metastasis control to cilia movement. Mol. Cell Biochem. 2009, 329, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.P.; Robinson, B.; Parks, R.E. Nucleoside diphosphokinase from erythrocytes. Methods Enzymol. 1978, 51, 376–386. [Google Scholar] [PubMed]
- Ma, D.; McCorkle, J.R.; Kaetzel, D.M. The metastasis suppressor NM23-H1 possesses 3′–5′ exonuclease activity. J. Biol. Chem. 2004, 279, 18073–18084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; McCorkle, J.R.; Novak, M.; Yang, M.; Kaetzel, D.M. Metastasis suppressor function of NM23-H1 requires its 3′–5′ exonuclease activity. Int. J. Cancer 2011, 128, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Jarrett, S.G.; Craven, R.; Kaetzel, D.M. YNK1, the yeast homolog of human metastasis suppressor NM23, is required for repair of UV radiation- and etoposide-induced DNA damage. Mutat. Res. 2009, 660, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Novak, M.; Dabernat, S.; Daniel, J.Y.; Mellon, I.; Zhang, Q.; Harris, N.; Ciesielski, M.J.; Fenstermaker, R.A.; Kovacic, D.; et al. Metastasis suppressor NM23-H1 promotes repair of UV-induced DNA damage and suppresses UV-induced melanomagenesis. Cancer Res. 2012, 72, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Cheng, L.; Xie, K.; Bucana, C.D.; Dong, Z. Direct correlation between DNA repair capacity and metastatic potential of K-1735 murine melanoma cells. J. Investig. Dermatol. 1997, 108, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann, A.; Rosselli, F.; Lazar, V.; Winnepenninckx, V.; Mansuet-Lupo, A.; Dessen, P.; van den Oord, J.J.; Spatz, A.; Sarasin, A. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2008, 27, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Jewell, R.; Conway, C.; Mitra, A.; Randerson-Moor, J.; Lobo, S.; Nsengimana, J.; Harland, M.; Marples, M.; Edward, S.; Cook, M.; et al. Patterns of expression of DNA repair genes and relapse from melanoma. Clin. Cancer Res. 2010, 16, 5211–5221. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yin, M.; Wang, L.-E.; Amos, C.I.; Zhu, D.; Lee, J.E.; Gershenwald, J.E.; Grimm, E.A.; Wei, Q. Polymorphisms of nucleotide excision repair genes predict melanoma survival. J. Investig. Dermatol. 2014, 133, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Sarasin, A.; Kauffmann, A. Overexpression of DNA repair genes is associated with metastasis: A new hypothesis. Mutat. Res. 2008, 659, 49–55. [Google Scholar] [CrossRef] [PubMed]
- McCorkle, J.R.; Leonard, M.K.; Kraner, S.D.; Blalock, E.M.; Ma, D.; Zimmer, S.G.; Kaetzel, D.M. The metastasis suppressor NME1 regulates expression of genes linked to metastasis and patient outcome in melanoma and breast carcinoma. Cancer Genom. Proteom. 2014, 11, 175–194. [Google Scholar]
- Horak, C.E.; Lee, J.H.; Elkahloun, A.G.; Boissan, M.; Dumont, S.; Maga, T.K.; Arnaud-Dabernat, S.; Palmieri, D.; Stetler-Stevenson, W.G.; Lacombe, M.L.; et al. Nm23-H1 suppresses tumor cell motility by down-regulating the lysophosphatidic acid receptor EDG2. Cancer Res. 2007, 67, 7238–7246. [Google Scholar] [CrossRef] [PubMed]
- Horak, C.E.; Mendoza, A.; Vega-Valle, E.; Albaugh, M.; Graff-Cherry, C.; McDermott, W.G.; Hua, E.; Merino, M.J.; Steinberg, S.M.; Khanna, C.; et al. Nm23-H1 suppresses metastasis by inhibiting expression of the lysophosphatidic acid receptor EDG2. Cancer Res. 2007, 67, 11751–11759. [Google Scholar] [CrossRef] [PubMed]
- Postel, E.H.; Berberich, S.J.; Flint, S.J.; Ferrone, C.A. Human c-myc transcription factor PuF identified as nm23-H2 nucleoside diphosphate kinase, a candidate suppressor of tumor metastasis. Science 1993, 261, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Xing, Z.; Liu, B.; Pedigo, N.; Zimmer, S.; Bai, Z.; Postel, E.; Kaetzel, D.M. NM23-H1 cleaves and represses transcriptional activity of nuclease-hypersensitive elements in the PDGF-A promoter. J. Biol. Chem. 2002, 277, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Postel, E.; Berberich, S.J.; Rooney, J.W.; Kaetzel, D.M. Human NM23/nucleoside diphosphate kinase regulates gene expression through DNA binding to nuclease-hypersensitive elements. J. Bioenerg. Biomembr. 2000, 32, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Cervoni, L.; Pietrangeli, P.; Chichiarelli, S.; Altieri, F.; Egistelli, L.; Turano, C.; Lascu, I.; Giartosio, A. In vivo cross-linking of nm23/nucleoside diphosphate kinase to the PDGF-A gene promoter. Mol. Biol. Rep. 2003, 30, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Cervoni, L.; Egistelli, L.; Eufemi, M.; d’Abusco, A.S.; Altieri, F.; Lascu, I.; Turano, C.; Giartosio, A. DNA sequences acting as binding sites for NM23/NDPK proteins in melanoma M14 cells. J. Cell Biochem. 2006, 98, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Moses, R.E.; O’Malley, B.W. DNA transcription and repair: A confluence. J. Biol. Chem. 2012, 287, 23266–23270. [Google Scholar] [CrossRef] [PubMed]
- Postel, E.H.; Wohlman, I.; Zou, X.; Juan, T.; Sun, N.; D’Agostin, D.; Cuellar, M.; Choi, T.; Notterman, D.A.; la Perle, K.M. Targeted deletion of Nm23/nucleoside diphosphate kinase A and B reveals their requirement for definitive erythropoiesis in the mouse embryo. Dev. Dyn. 2009, 238, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Postel, E.H.; Zou, X.; Notterman, D.A.; la Perle, K.M. Double knockout Nme1/Nme2 mouse model suggests a critical role for NDP kinases in erythroid development. Mol. Cell Biochem. 2009, 329, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Arnaud-Dabernat, S.; Bourbon, P.M.; Dierich, A.; le Meur, M.; Daniel, J.-Y. Knockout mice as model systems for studying nm23/NDP kinase gene functions. Application to the nm23-M1 gene. J. Bioenerg. Biomembr. 2003, 35, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Srivastava, S.; Zhdanova, O.; Sun, Y.; Li, Z.; Skolnik, E.Y. Nucleoside diphosphate kinase B knock-out mice have impaired activation of the K+ channel KCa3.1, resulting in defective T cell activation. J. Biol. Chem. 2010, 285, 38765–38771. [Google Scholar] [PubMed]
- Ha, L.; Ichikawa, T.; Anver, M.; Dickins, R.; Lowe, S.; Sharpless, N.E.; Krimpenfort, P.; DePinho, R.A.; Bennett, D.C.; Sviderskaya, E.V.; et al. ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc. Natl. Acad. Sci. USA 2007, 104, 10968–10973. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Hornyak, T.J.; Merlino, G. A genetically engineered mouse model with inducible GFP expression in melanocytes. Pigment Cell Melanoma Res. 2011, 24, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Davis, S.; Noonan, F.P.; Graff-Cherry, C.; Hawley, T.S.; Walker, R.L.; Feigenbaum, L.; Fuchs, E.; Lyakh, L.; Young, H.A.; et al. Interferon-γ links ultraviolet radiation to melanomagenesis in mice. Nature 2011, 469, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Foster, P.J.; Dunn, E.A.; Karl, K.E.; Snir, J.A.; Nycz, C.M.; Harvey, A.J.; Pettis, R.J. Cellular magnetic resonance imaging: In vivo imaging of melanoma cells in lymph nodes of mice. Neoplasia 2008, 10, 207–216. [Google Scholar]
- Puaux, A.L.; Ong, L.C.; Jin, Y.; Teh, I.; Hong, M.; Chow, P.K.; Golay, X.; Abastado, J.P. A comparison of imaging techniques to monitor tumor growth and cancer progression in living animals. Int. J. Mol. Imaging 2011, 2011, 321538. [Google Scholar] [CrossRef] [PubMed]
- Noonan, F.P.; Otsuka, T.; Bang, S.; Anver, M.R.; Merlino, G. Accelerated ultraviolet radiation-induced carcinogenesis in hepatocyte growth factor/scatter factor transgenic mice. Cancer Res. 2000, 60, 3738–3743. [Google Scholar] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Tas, F. Metastatic behavior in melanoma: Timing, pattern, survival, and influencing factors. J. Oncol. 2012, 2012, 647684. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. BrafV600E cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Kircher, D.A.; Arave, R.A.; Cho, J.H.; Holmen, S.L. Melanoma metastases caught in the AKT. Mol. Cell Oncol. 2016, 3, e1128516. [Google Scholar] [CrossRef] [PubMed]
- Ha, L.; Noonan, F.P.; de Fabo, E.C.; Merlino, G. Animal models of melanoma. J. Investig. Dermatol. Symp. Proc. 2005, 10, 86–88. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leonard, M.K.; Pamidimukkala, N.; Puts, G.S.; Snyder, D.E.; Slominski, A.T.; Kaetzel, D.M. The HGF/SF Mouse Model of UV-Induced Melanoma as an In Vivo Sensor for Metastasis-Regulating Gene. Int. J. Mol. Sci. 2017, 18, 1647. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081647
Leonard MK, Pamidimukkala N, Puts GS, Snyder DE, Slominski AT, Kaetzel DM. The HGF/SF Mouse Model of UV-Induced Melanoma as an In Vivo Sensor for Metastasis-Regulating Gene. International Journal of Molecular Sciences. 2017; 18(8):1647. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081647
Chicago/Turabian StyleLeonard, M. Kathryn, Nidhi Pamidimukkala, Gemma S. Puts, Devin E. Snyder, Andrzej T. Slominski, and David M. Kaetzel. 2017. "The HGF/SF Mouse Model of UV-Induced Melanoma as an In Vivo Sensor for Metastasis-Regulating Gene" International Journal of Molecular Sciences 18, no. 8: 1647. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081647