Establishing Computational Approaches Towards Identifying Malarial Allosteric Modulators: A Case Study of Plasmodium falciparum Hsp70s

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
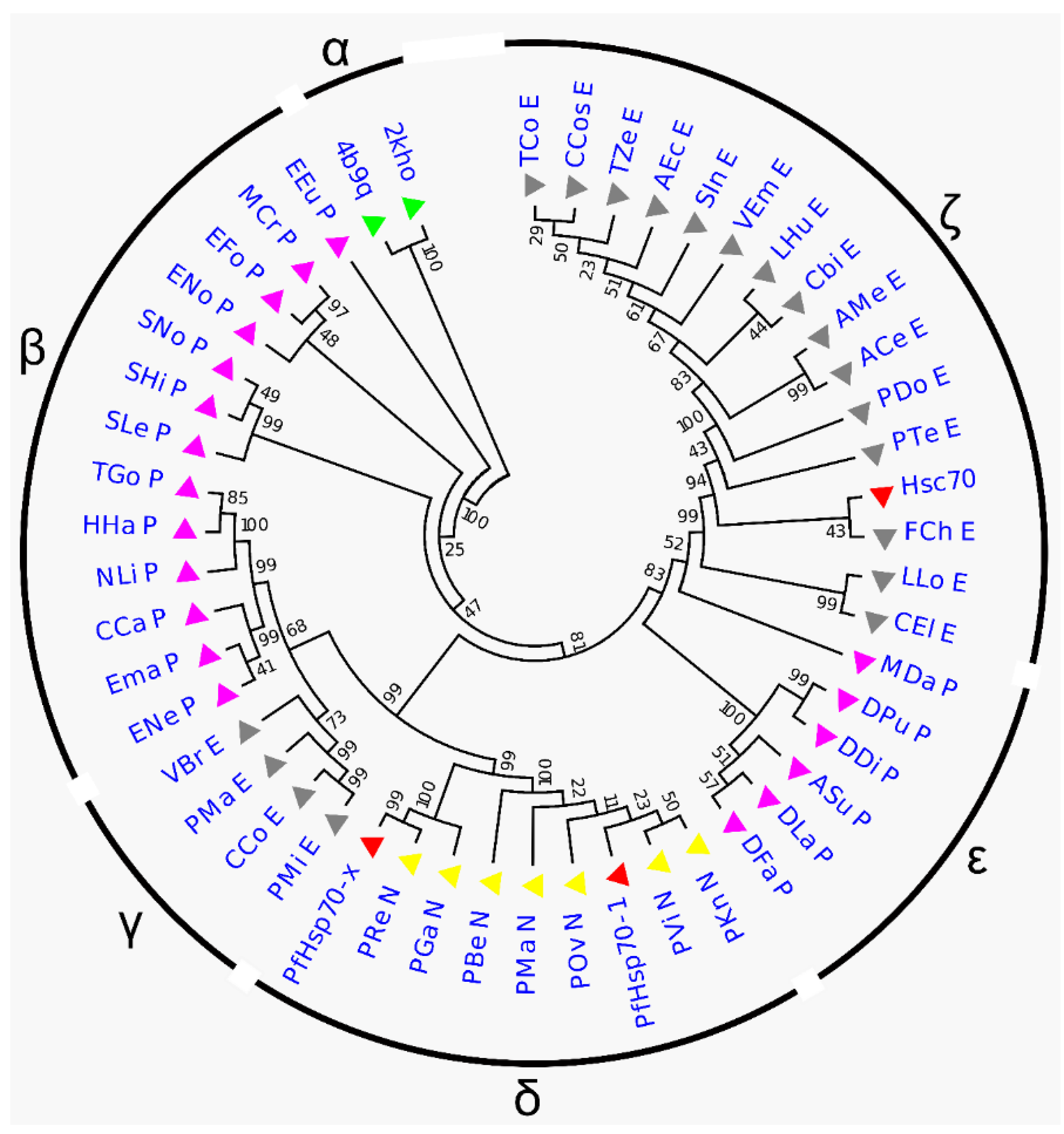
2.1. Phylogenetic Tree Analysis
2.2. Homology Modeling
2.3. Molecular Docking
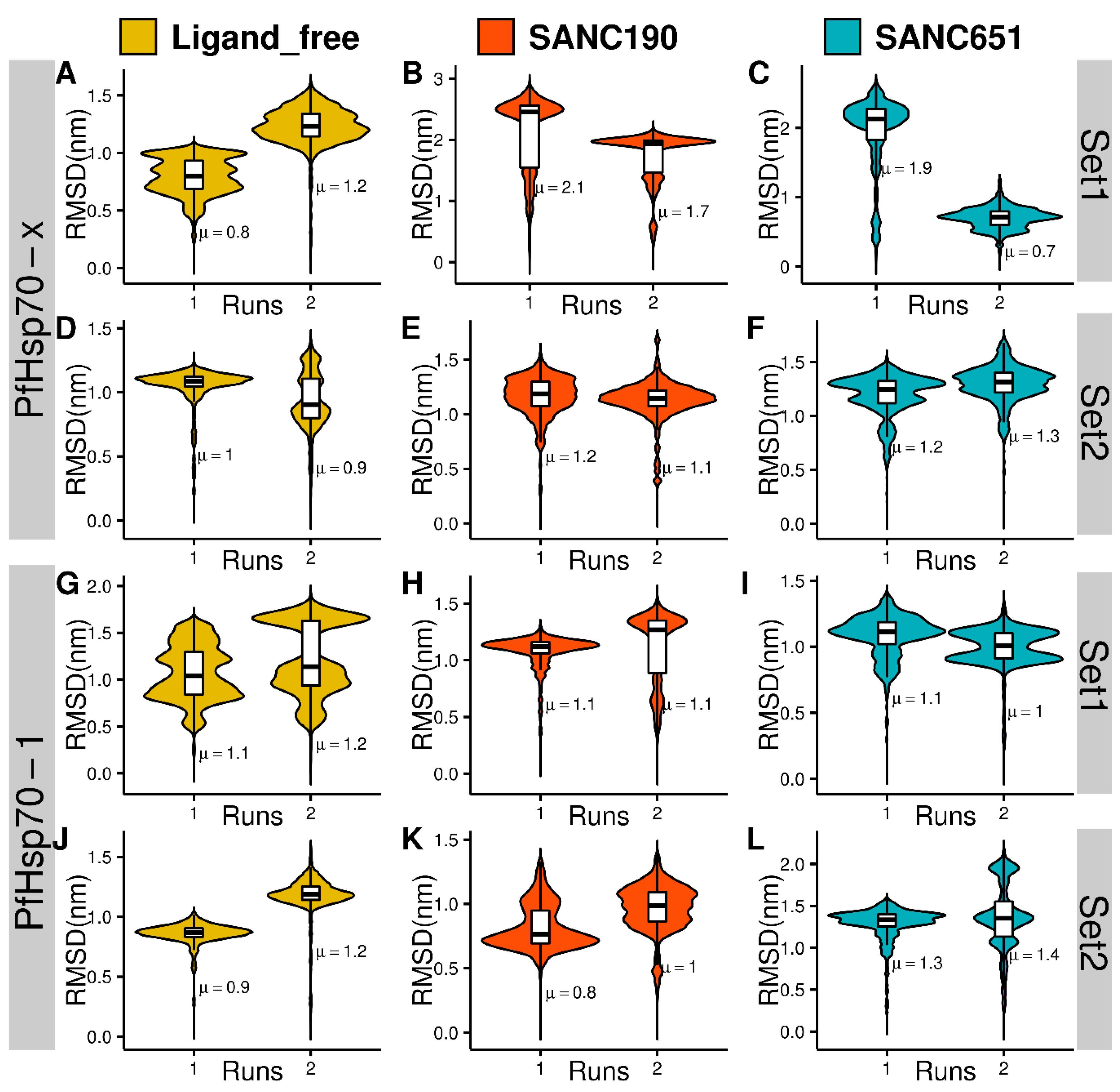
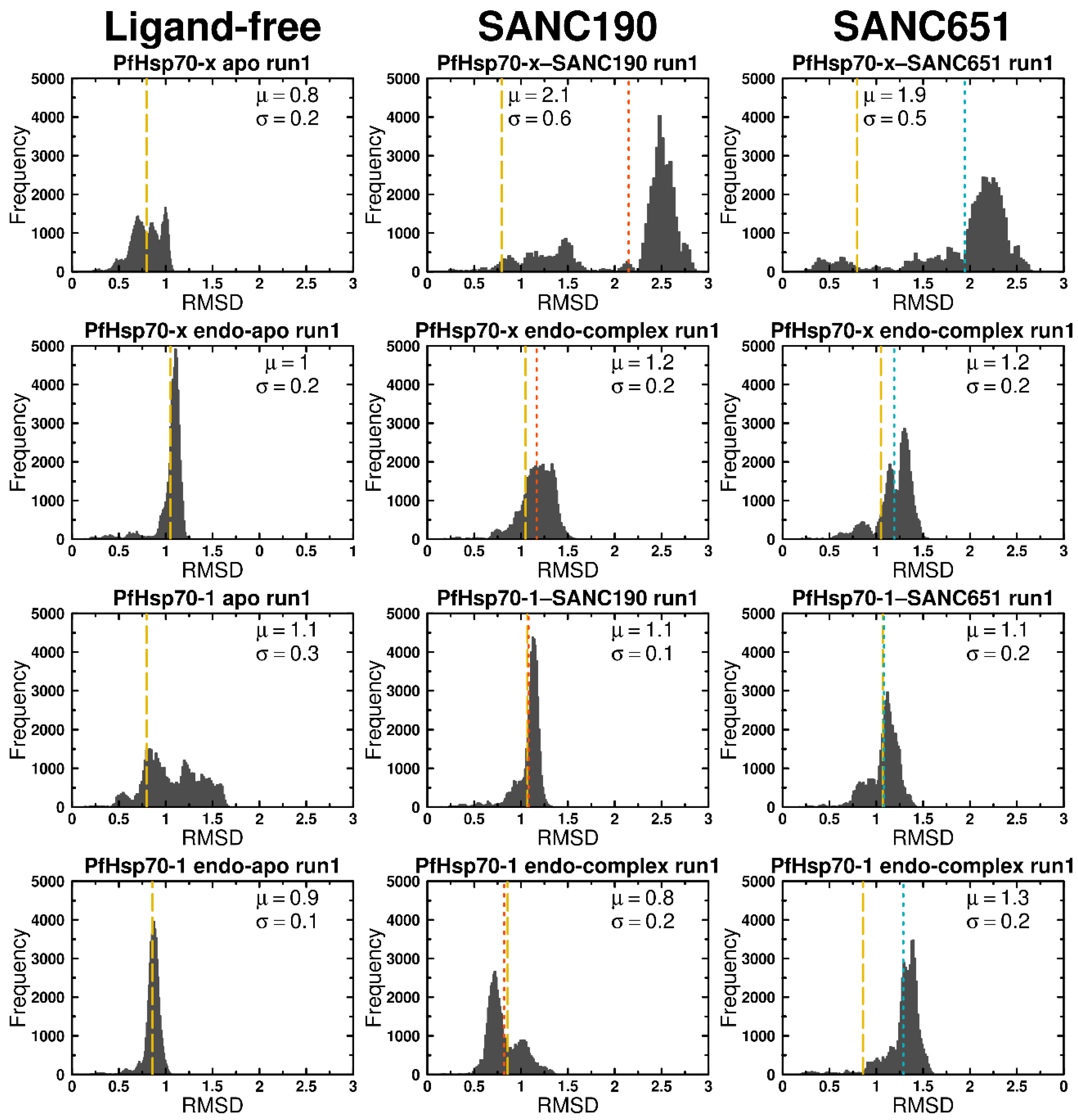
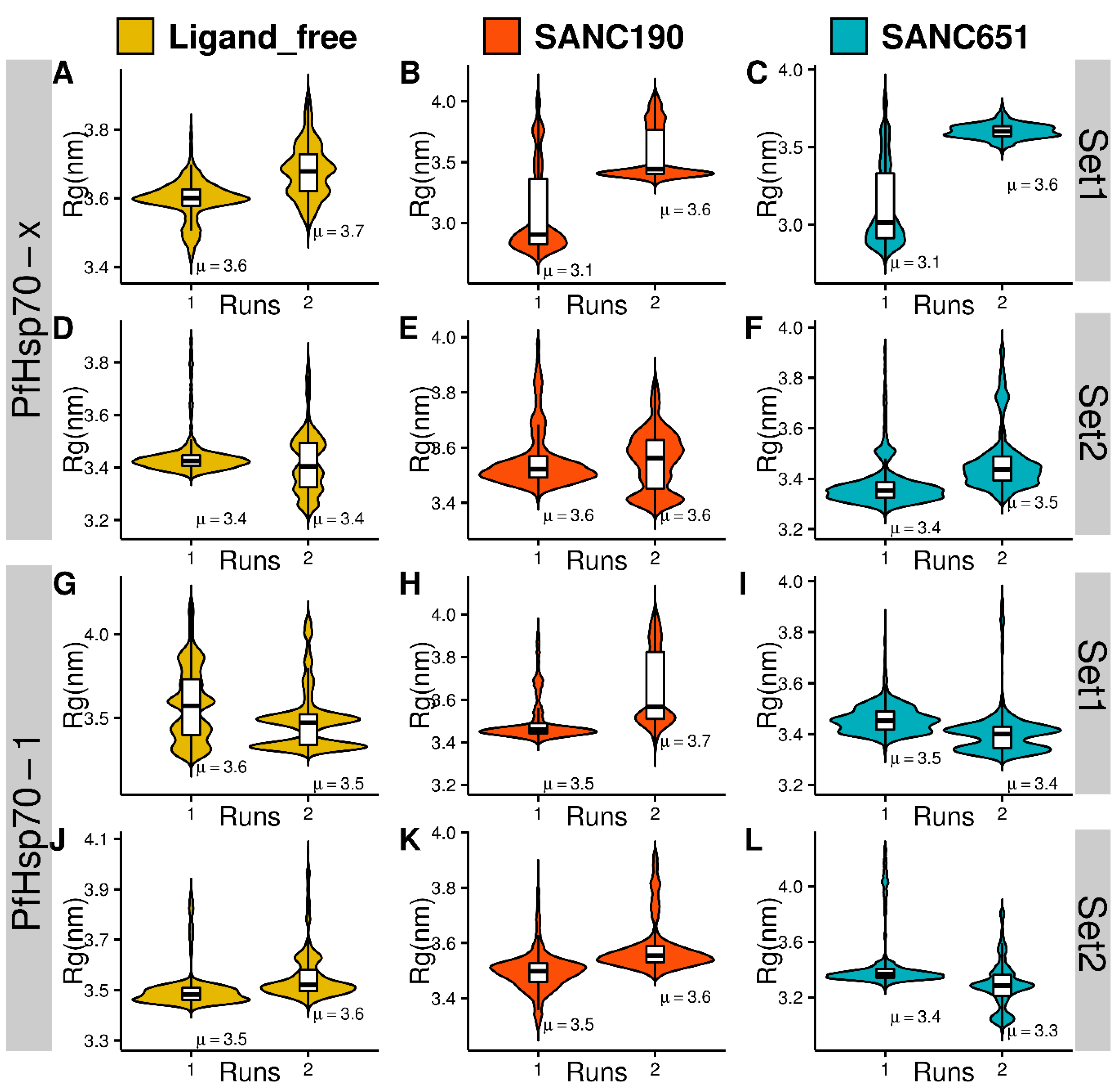
2.4. Molecular Dynamics
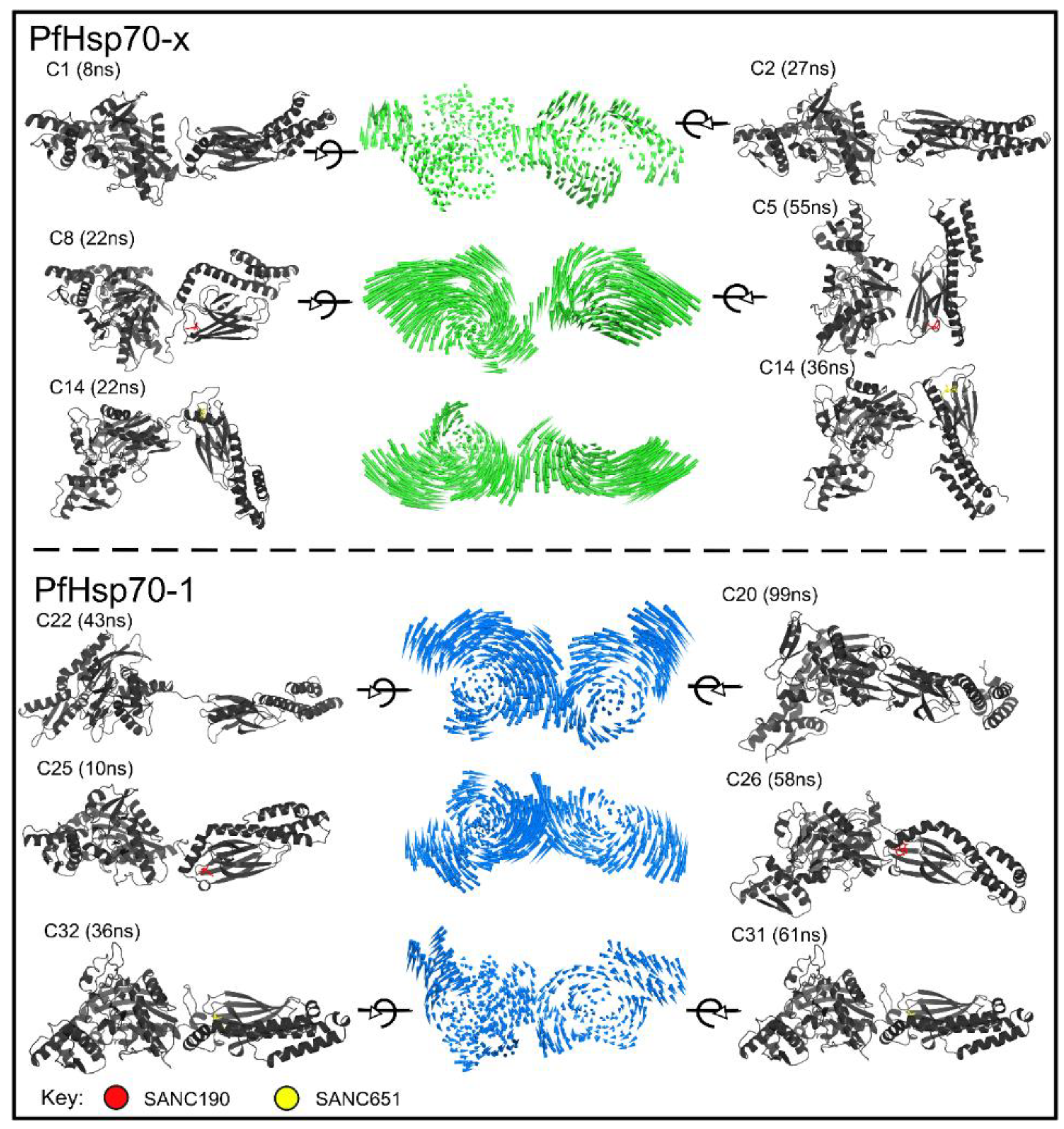
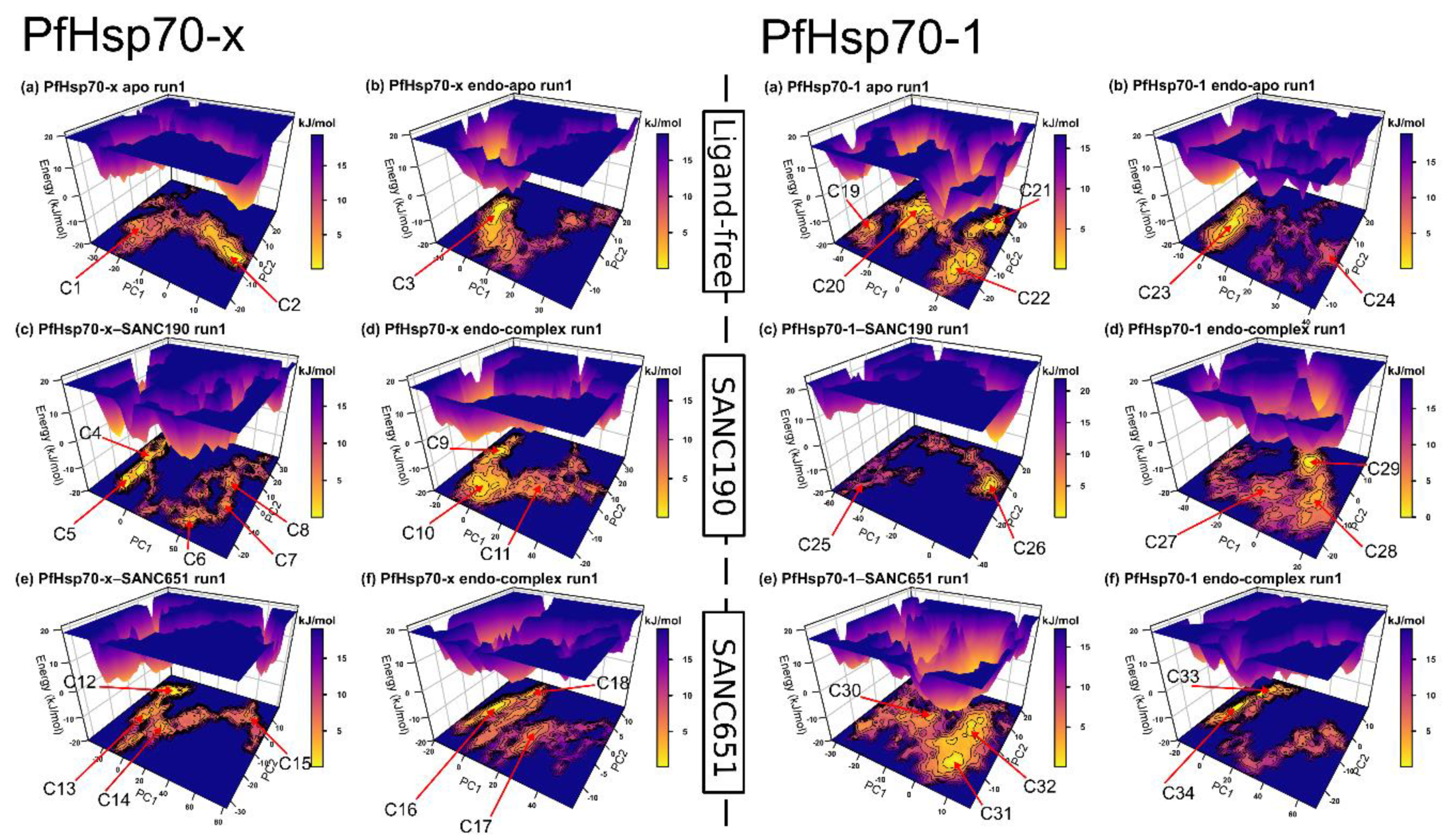
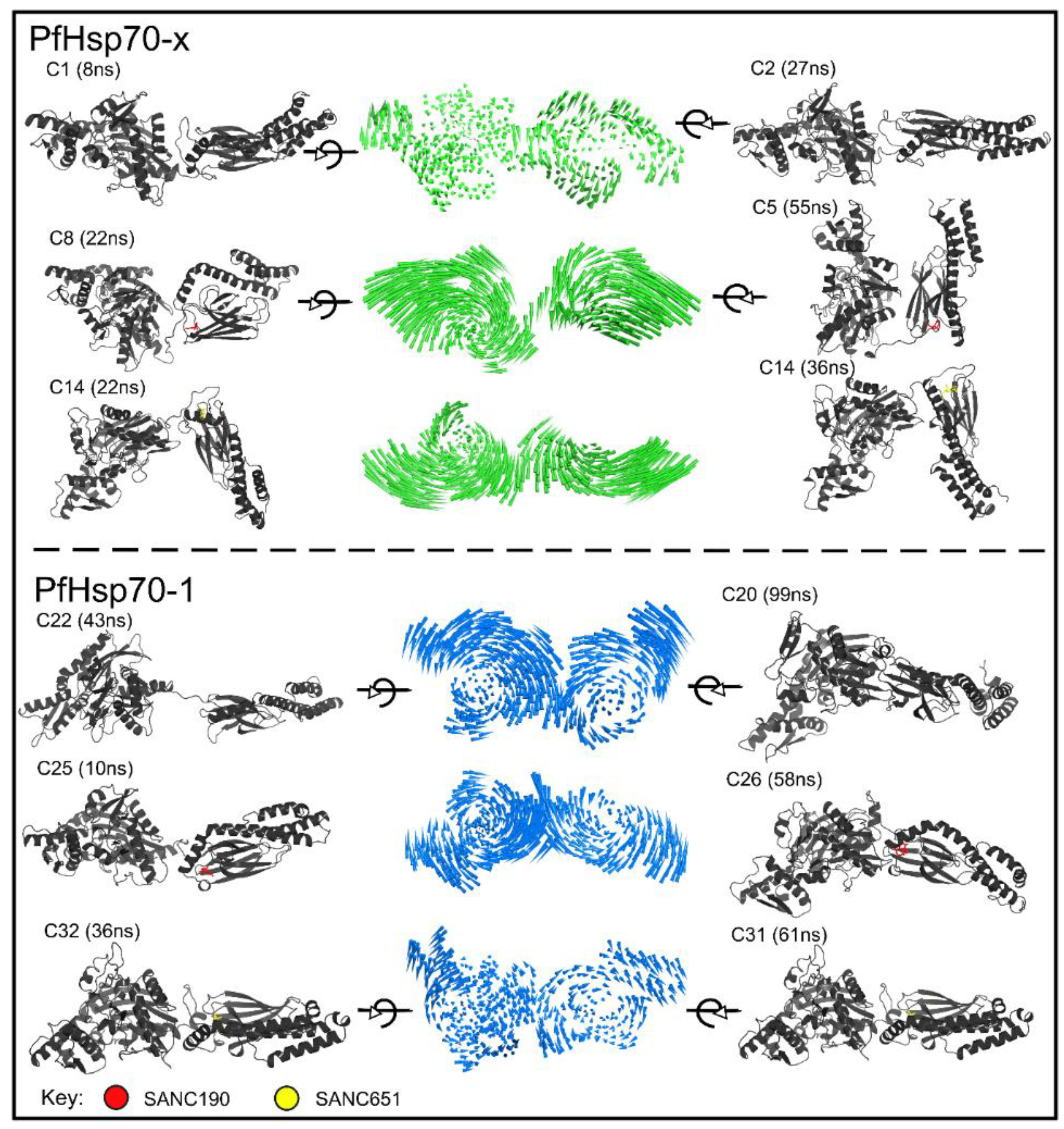
2.5. Essential Dynamics
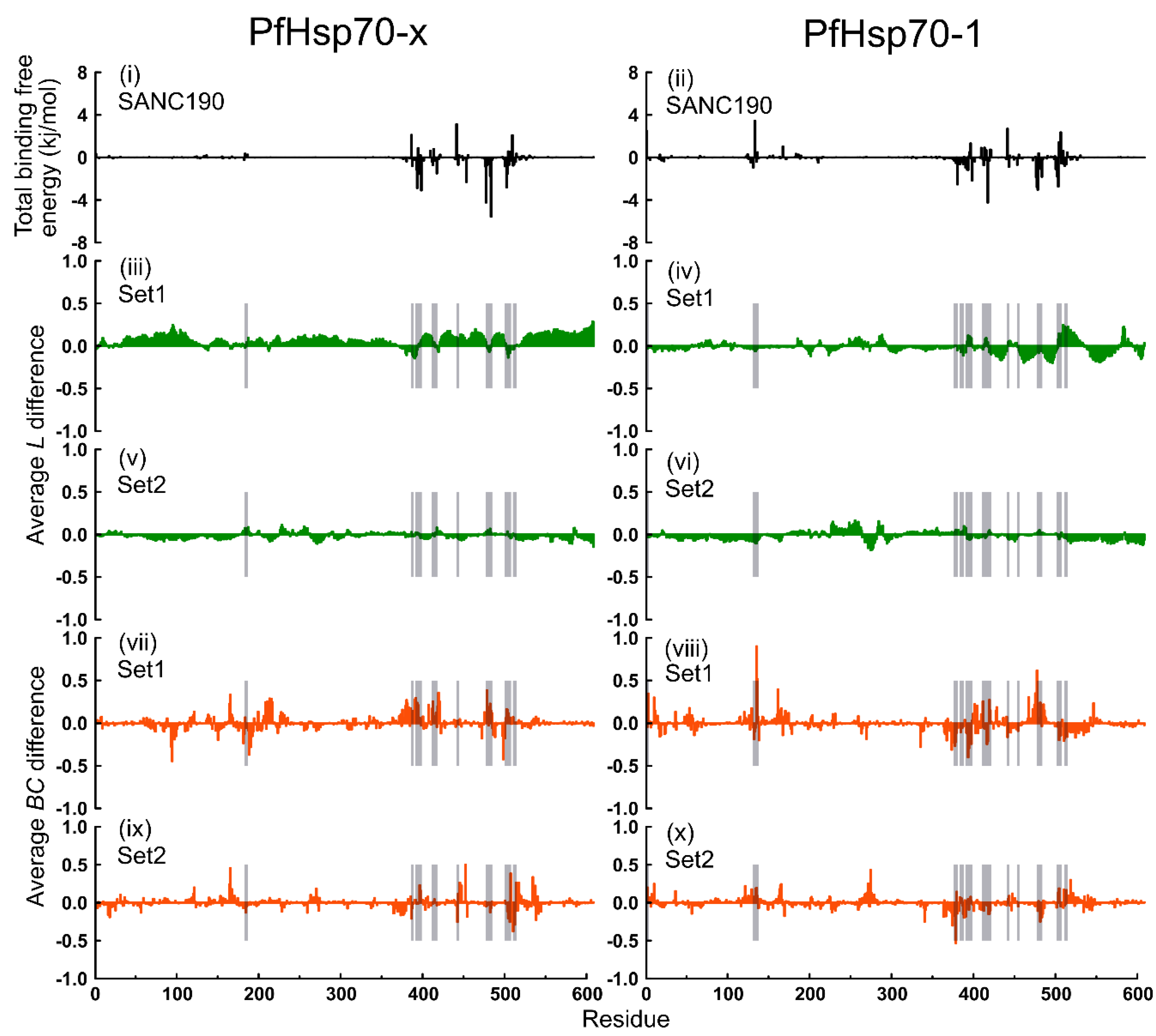
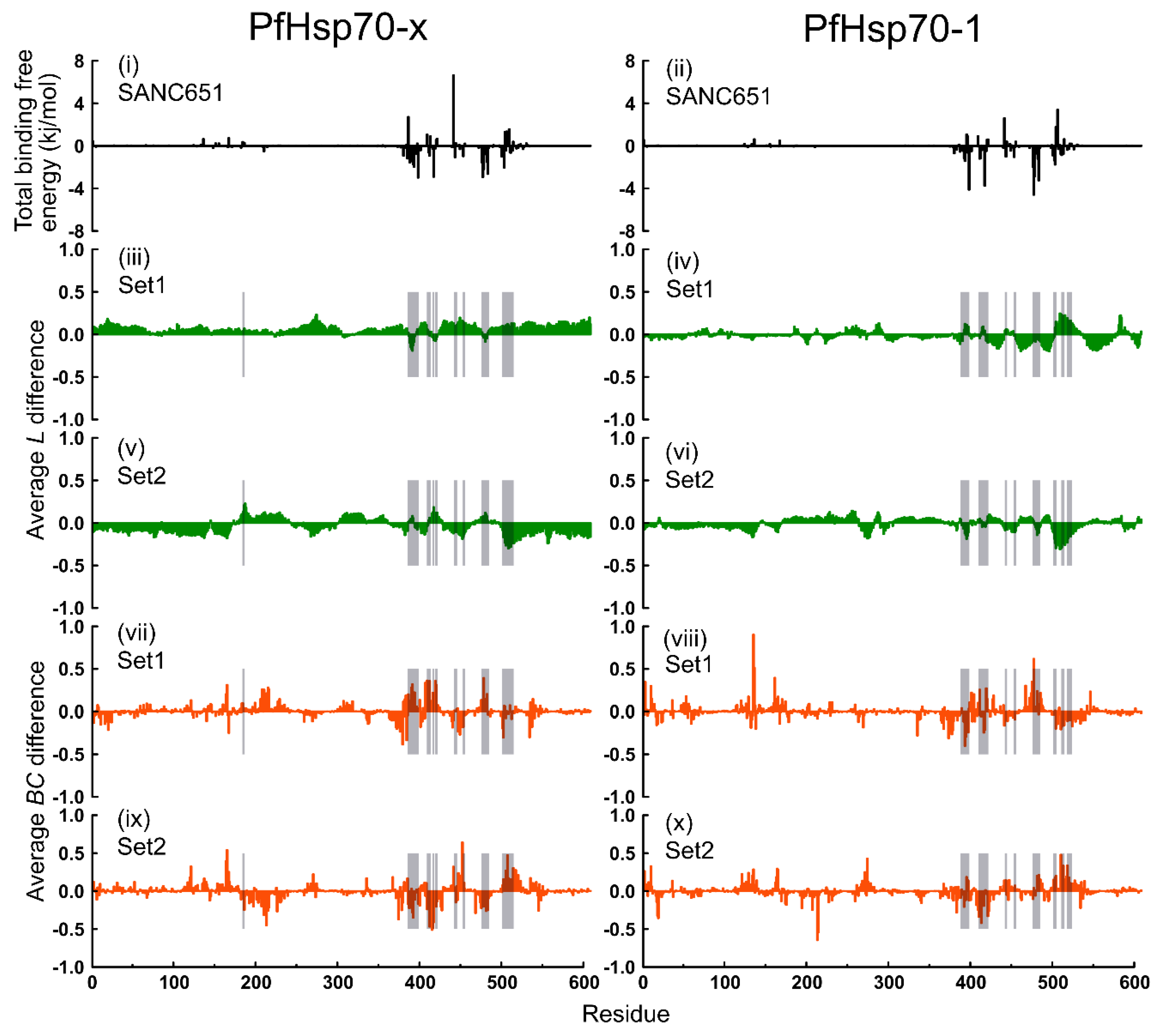
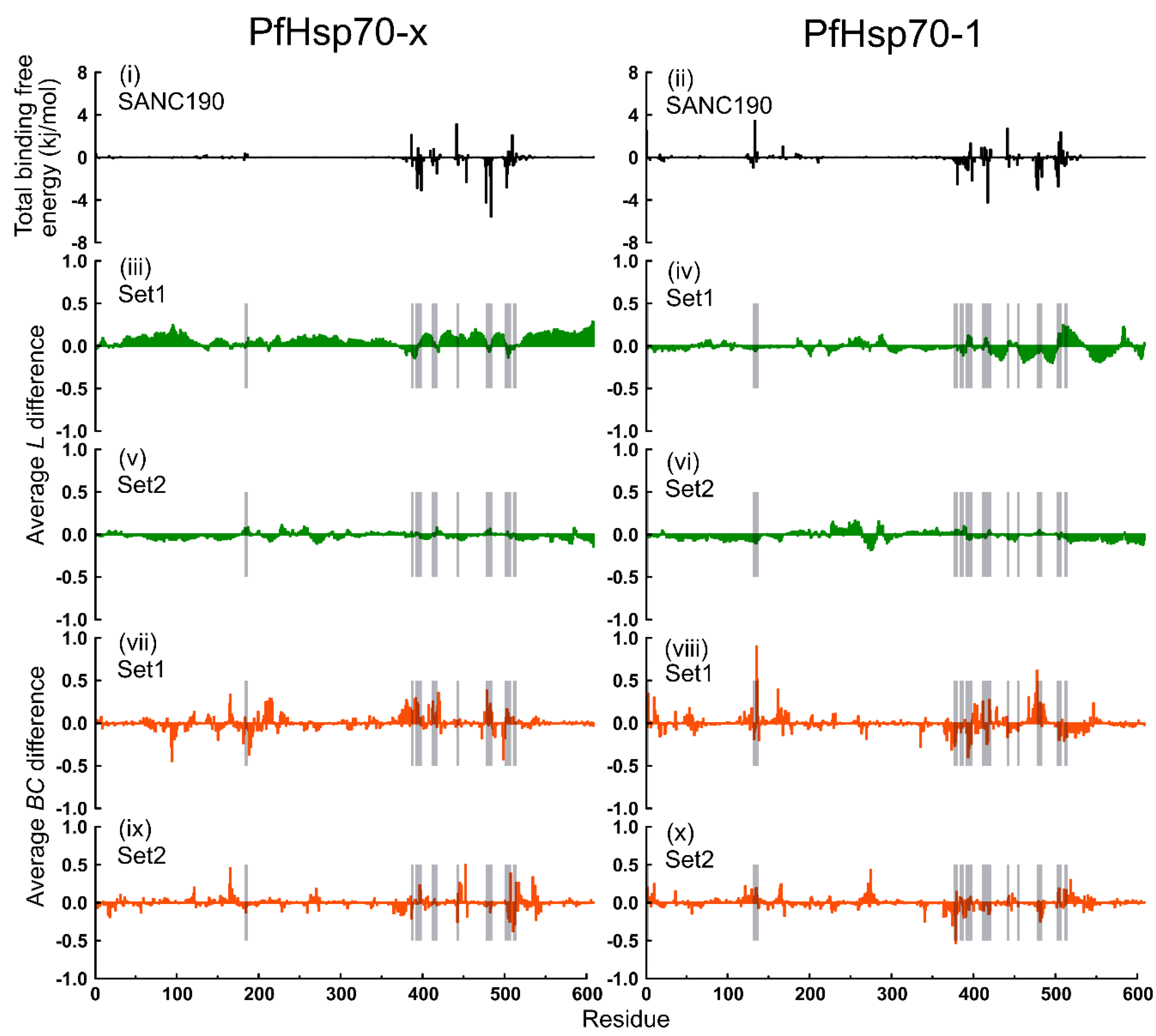
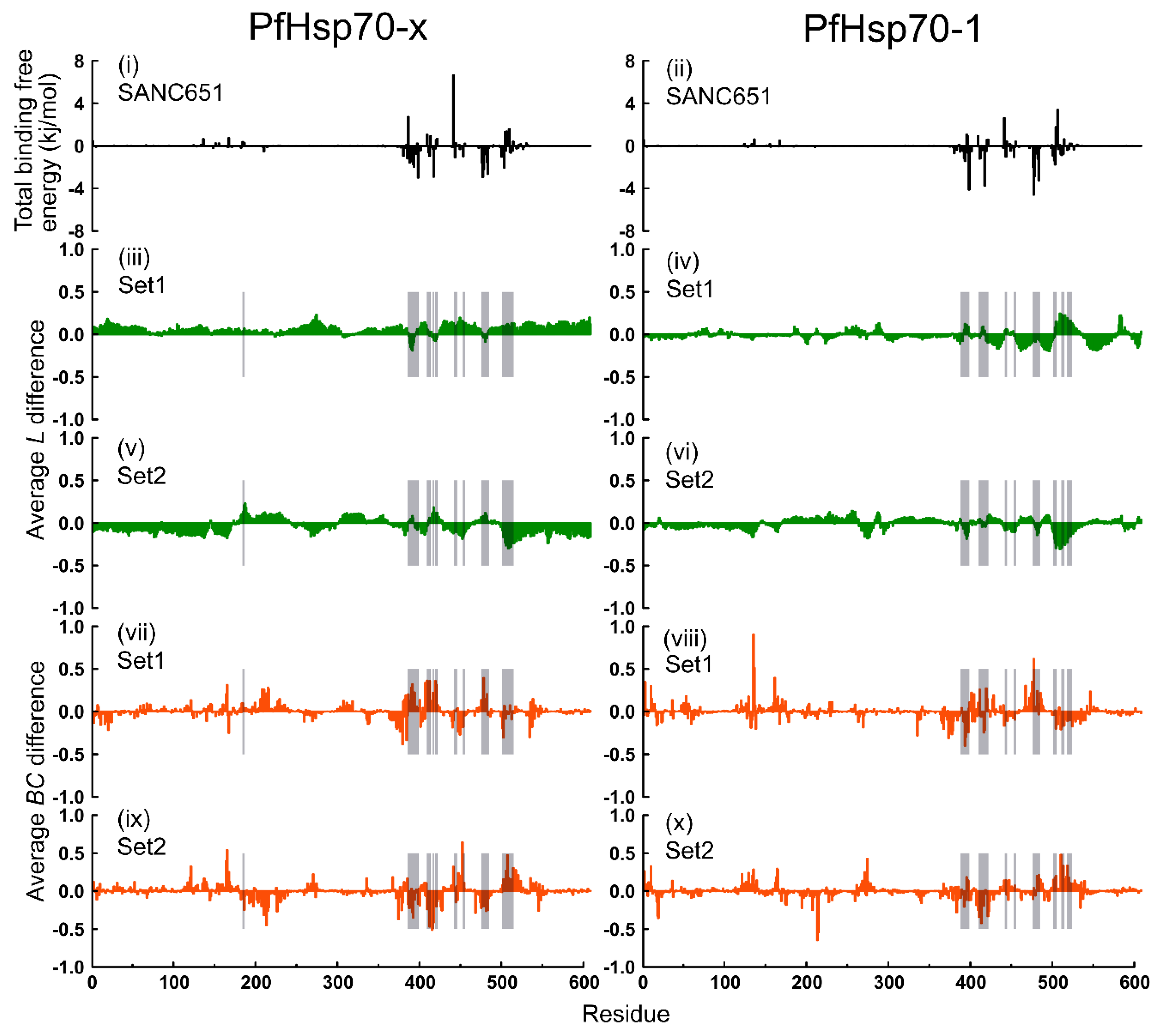
2.6. Thermodynamic Assessment
2.7. Dynamic Residue Network Analysis
3. Materials and Methods
3.1. Data Retrieval and Phylogenetic Tree Analysis
3.2. Homology Modeling
3.3. Molecular Docking
3.4. All Atom Molecular Dynamics Simulations
3.5. Coarse-Grained Monte Carlo Dynamics Simulations
3.6. Preliminary Analysis of Trajectories
3.7. Essential Dynamics
3.8. Binding Free Energy Analysis
3.9. Dynamic Residue Network Analysis
3.10. Coarse-Grained Dynamic Residue Network Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Organization, W.H. World Malaria Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Witkowski, B.; Lelièvre, J.; Barragán, M.J.L.; Laurent, V.; Su, X.; Berry, A.; Benoit-Vical, F. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob. Agents Chemother. 2010, 54, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Musyoka, T.M.; Kanzi, A.M.; Lobb, K.A.; Tastan Bishop, Ö. Structure Based Docking and Molecular Dynamic Studies of Plasmodial Cysteine Proteases against a South African Natural Compound and its Analogs. Sci. Rep. 2016, 6, 23690. [Google Scholar] [CrossRef]
- Musyoka, T.M.; Kanzi, A.M.; Lobb, K.A.; Tastan Bishop, Ö. Analysis of non-peptidic compounds as potential malarial inhibitors against Plasmodial cysteine proteases via integrated virtual screening workflow. J. Biomol. Struct. Dyn. 2016, 34, 2084–2101. [Google Scholar] [CrossRef]
- Mugumbate, G.; Newton, A.S.; Rosenthal, P.J.; Gut, J.; Moreira, R.; Chibale, K.; Guedes, R.C. Novel anti-Plasmodial hits identified by virtual screening of the ZINC database. J. Comput. Aided. Mol. Des. 2013. [Google Scholar] [CrossRef]
- Blundell, T.L.; Sibanda, B.L.; Montalvão, R.W.; Brewerton, S.; Chelliah, V.; Worth, C.L.; Harmer, N.J.; Davies, O.; Burke, D. Structural biology and bioinformatics in drug design: Opportunities and challenges for target identification and lead discovery. Philos. Trans. R. Soc. B Biol. Sci. 2006. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.; D’Annessa, I.; Moroni, E.; Morra, G.; Paladino, A.; Rinaldi, S.; Compostella, F.; Colombo, G. Allosteric Modulators of HSP90 and HSP70: Dynamics Meets Function through Structure-Based Drug Design. J. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Shrestha, L.; Bolaender, A.J.; Patel, H.; Taldone, T. Heat Shock Protein (HSP) Drug Discovery and Development: Targeting Heat Shock Proteins in Disease. Curr. Top. Med. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Howe, M.K.; Speer, B.L.; Hughes, P.F.; Loiselle, D.R.; Vasudevan, S.; Haystead, T.A.J. An inducible heat shock protein 70 small molecule inhibitor demonstrates anti-dengue virus activity, validating Hsp70 as a host antiviral target. Antivir. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–580. [Google Scholar] [CrossRef]
- Penkler, D.; Sensoy, Ö.; Atilgan, C.; Tastan Bishop, Ö. Perturbation-Response Scanning Reveals Key Residues for Allosteric Control in Hsp70. J. Chem. Inf. Model. 2017, 57, 1359–1374. [Google Scholar] [CrossRef]
- English, C.A.; Sherman, W.; Meng, W.; Gierasch, L.M. The Hsp70 interdomain linker is a dynamic switch that enables allosteric communication between two structured domains. J. Biol. Chem. 2017, 292, 14765–14774. [Google Scholar] [CrossRef] [Green Version]
- Zuiderweg, E.R.P.; Bertelsen, E.B.; Rousaki, A.; Mayer, M.P.; Gestwicki, J.E.; Ahmad, A. Allostery in the Hsp70 chaperone proteins. Top. Curr. Chem. 2013, 328, 99–153. [Google Scholar] [CrossRef]
- Zhuravleva, A.; Clerico, E.M.; Gierasch, L.M. An Interdomain Energetic Tug-of-War Creates the Allosterically Active State in Hsp70 Molecular Chaperones. Cell 2012, 151, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Zhuravleva, A.; Gierasch, L.M. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc. Natl. Acad. Sci. USA 2011, 108, 6987–6992. [Google Scholar] [CrossRef] [Green Version]
- Bertelsen, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef]
- Mayer, M.P. Intra-molecular pathways of allosteric control in Hsp70s. Philos. Trans. R. Soc. B Biol. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, A.; Gierasch, L.M. Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc. Natl. Acad. Sci. USA 2015, 112, E2865–E2873. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.; Kumar, R.; Tatu, U. Chaperoning a cellular upheaval in malaria: Heat shock proteins in Plasmodium falciparum. Mol. Biochem. Parasitol. 2007, 153, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Hatherley, R.; Blatch, G.L.; Bishop, O.T. Plasmodium falciparum Hsp70-x: A heat shock protein at the host-parasite interface. J. Biomol. Struct. Dyn. 2014, 32, 1766–1779. [Google Scholar] [CrossRef] [PubMed]
- Sargeant, T.J.; Marti, M.; Caler, E.; Carlton, J.M.; Simpson, K.; Speed, T.P.; Cowman, A.F. Lineage-specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol. 2006, 7, R12. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.; Pallavi, R.; Chandran, S.; Chakravarti, H.; Middha, S.; Acharya, J.; Kochar, S.; Kochar, D.; Subudhi, A.; Boopathi, A.P.; et al. A glimpse into the clinical proteome of human malaria parasites Plasmodium falciparum and Plasmodium vivax. Proteom. Clin. Appl. 2009. [Google Scholar] [CrossRef] [PubMed]
- Grover, M.; Chaubey, S.; Ranade, S.; Tatu, U. Identification of an exported heat shock protein 70 in Plasmodium falciparum. Parasite 2013, 20, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, B.; Biswas, S.; Sharma, Y.D. Effect of heat-shock on Plasmodium falciparum viability, growth and expression of the heat-shock protein “PFHSP70-I” gene. FEBS Lett. 1992. [Google Scholar] [CrossRef]
- Cabral, F.J.; Vianna, L.G.; Medeiros, M.M.; Carlos, B.C.; Martha, R.D.; Silva, N.M.; Da Silva, L.H.P.; Stabeli, R.G.; Wunderlich, G. Immunoproteomics of Plasmodium falciparum-infected red blood cell membrane fractions. Mem. Inst. Oswaldo Cruz 2017. [Google Scholar] [CrossRef]
- Chiang, A.N.; Valderramos, J.-C.; Balachandran, R.; Chovatiya, R.J.; Mead, B.P.; Schneider, C.; Bell, S.L.; Klein, M.G.; Huryn, D.M.; Chen, X.S.; et al. Select pyrimidinones inhibit the propagation of the malarial parasite, Plasmodium falciparum. Bioorg. Med. Chem. 2009, 17, 1527–1533. [Google Scholar] [CrossRef] [Green Version]
- Freeman, B.C.; Myers, M.P.; Schumacher, R.; Morimoto, R.I. Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. EMBO J. 1995. [Google Scholar] [CrossRef]
- Ramya, T.N.C.; Surolia, N.; Surolia, A. 15-Deoxyspergualin modulates Plasmodium falciparum heat shock protein function. Biochem. Biophys. Res. Commun. 2006, 348, 585–592. [Google Scholar] [CrossRef]
- Pesce, E.-R.; Cockburn, I.L.; Goble, J.L.; Stephens, L.L.; Blatch, G.L. Malaria Heat Shock Proteins: Drug Targets that Chaperone other Drug Targets. Infect. Disord. Drug Targets 2012, 10, 147–157. [Google Scholar] [CrossRef]
- Przyborski, J.M.; Diehl, M.; Blatch, G.L. Plasmodial HSP70s are functionally adapted to the malaria parasite life cycle. Front. Mol. Biosci. 2015, 2, 34. [Google Scholar] [CrossRef] [Green Version]
- Sharma, Y.D. Structure and possible function of heat-shock proteins in falciparum malaria. Comp. Biochem. Physiol. Part B Comp. Biochem. 1992, 102, 437–444. [Google Scholar] [CrossRef]
- Biswas, S.; Sharma, Y.D. Enhanced expression of Plasmodium falciparum heat shock protein PFHSP70-I at higher temperatures and parasite survival. FEMS Microbiol. Lett. 1994, 124, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Natalang, O.; Bischoff, E.; Deplaine, G.; Proux, C.; Dillies, M.-A.; Sismeiro, O.; Guigon, G.; Bonnefoy, S.; Patarapotikul, J.; Mercereau-Puijalon, O.; et al. Dynamic RNA profiling in Plasmodium falciparum synchronized blood stages exposed to lethal doses of artesunate. BMC Genom. 2008, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Mabate, B.; Zininga, T.; Ramatsui, L.; Makumire, S.; Achilonu, I.; Dirr, H.W.; Shonhai, A. Structural and biochemical characterization of Plasmodium falciparum Hsp70-x reveals functional versatility of its C-terminal EEVN motif. Proteins Struct. Funct. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Charnaud, S.C.; Dixon, M.W.A.; Nie, C.Q.; Chappell, L.; Sanders, P.R.; Nebl, T.; Hanssen, E.; Berriman, M.; Chan, J.A.; Blanch, A.J.; et al. The exported chaperone Hsp70-x supports virulence functions for Plasmodium falciparum blood stage parasites. PLoS ONE 2017. [Google Scholar] [CrossRef]
- Cobb, D.W.; Florentin, A.; Fierro, M.A.; Krakowiak, M.; Moore, J.M.; Muralidharan, V. The Exported Chaperone PfHsp70x Is Dispensable for the Plasmodium falciparum Intraerythrocytic Life Cycle. mSphere 2017. [Google Scholar] [CrossRef]
- Rist, W.; Graf, C.; Bukau, B.; Mayer, M.P. Amide hydrogen exchange reveals conformational changes in Hsp70 chaperones important for allosteric regulation. J. Biol. Chem. 2006. [Google Scholar] [CrossRef]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015. [Google Scholar] [CrossRef]
- Jones, A.M.; Westwood, I.M.; Osborne, J.D.; Matthews, T.P.; Cheeseman, M.D.; Rowlands, M.G.; Jeganathan, F.; Burke, R.; Lee, D.; Kadi, N.; et al. A fragment-based approach applied to a highly flexible target: Insights and challenges towards the inhibition of HSP70 isoforms. Sci. Rep. 2016, 6, 34701. [Google Scholar] [CrossRef]
- Pettinger, J.; Le Bihan, Y.-V.; Widya, M.; van Montfort, R.L.M.; Jones, K.; Cheeseman, M.D. An Irreversible Inhibitor of HSP72 that Unexpectedly Targets Lysine-56. Angew. Chem. Int. Ed. Engl. 2017, 56, 3536–3540. [Google Scholar] [CrossRef]
- Leu, J.I.-J.; Zhang, P.; Murphy, M.E.; Marmorstein, R.; George, D.L. Structural basis for the inhibition of HSP70 and DnaK chaperones by small-molecule targeting of a C-terminal allosteric pocket. ACS Chem. Biol. 2014, 9, 2508–2516. [Google Scholar] [CrossRef]
- Penkler, D.; Bishop, O.T. Modulation of Human Hsp90α Conformational Dynamics by Allosteric Ligand Interaction at the C-Terminal Domain. bioRxiv 2018, 386755. [Google Scholar] [CrossRef] [PubMed]
- Amusengeri, A.; Bishop, Ö.T. Discorhabdin N, a South African natural compound, for Hsp72 and Hsc70 allosteric modulation: Combined study of molecular modeling and dynamic residue network analysis. Molecules 2019, 24, 188. [Google Scholar] [CrossRef] [PubMed]
- D’Annessa, I.; Raniolo, S.; Limongelli, V.; Di Marino, D.; Colombo, G. Ligand Binding, Unbinding and Allosteric Effects: Deciphering Small-Molecule Modulation of HSP90. J. Chem. Theory Comput. 2019. [Google Scholar] [CrossRef] [PubMed]
- Penkler, D.; Atilgan, C.; Tastan Bishop, Ö. Allosteric Modulation of Human Hsp90α Conformational Dynamics. J. Chem. Inf. Model. 2018, 58, 383–404. [Google Scholar] [CrossRef]
- Külzer, S.; Charnaud, S.; Dagan, T.; Riedel, J.; Mandal, P.; Pesce, E.R.; Blatch, G.L.; Crabb, B.S.; Gilson, P.R.; Przyborski, J.M. Plasmodium falciparum-encoded exported hsp70/hsp40 chaperone/co-chaperone complexes within the host erythrocyte. Cell. Microbiol. 2012, 14, 1784–1795. [Google Scholar] [CrossRef]
- Hatherley, R.; Brown, D.K.; Glenister, M.; Tastan Bishop, Ö. PRIMO: An Interactive Homology Modeling Pipeline. PLoS ONE 2016, 11, e0166698. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [Green Version]
- de Beer, T.A.P.; Joubert, F.; South African Association for the Advancement of Science. South African Journal of Science; South African Association for the Advancement of Science: Pretoria, South Africa, 2008; Volume 104. [Google Scholar]
- Šali, A. MODELLER: A Program for Protein Structure Modeling Release 9.12, r9480; The Rockefeller University: New York, NY, USA, 2013; pp. 779–815. [Google Scholar]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. IUCr PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W.L. PyMOL. Available online: https://www.pymol.org/ (accessed on 6 October 2019).
- Rodina, A.; Patel, P.D.; Kang, Y.; Patel, Y.; Baaklini, I.; Wong, M.J.H.; Taldone, T.; Yan, P.; Yang, C.; Maharaj, R.; et al. Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein. Chem. Biol. 2013, 20, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Leu, J.I.-J.; Murphy, M.E.; George, D.L.; Marmorstein, R. Crystal Structure of the Stress-Inducible Human Heat Shock Protein 70 Substrate-Binding Domain in Complex with Peptide Substrate. PLoS ONE 2014, 9, e103518. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P.; Hersey, A.; Montanari, D.; Overington, J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discov. 2011, 10, 197–208. [Google Scholar] [CrossRef]
- Huggins, D.J.; Sherman, W.; Tidor, B. Rational approaches to improving selectivity in drug design. J. Med. Chem. 2012, 55, 1424–1444. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- McPhail, K.L.; Davies-Coleman, M.T.; Starmer, J. Sequestered chemistry of the Arminacean nudibranch Leminda millecra in Algoa Bay, South Africa. J. Nat. Prod. 2001, 64, 1183–1190. [Google Scholar] [CrossRef]
- Mashimbye, M.J.; Maumela, M.C.; Drewes, S.E. A drimane sesquiterpenoid lactone from Warburgia salutaris. Phytochemistry 1999, 51, 435–438. [Google Scholar] [CrossRef]
- Leonard, C.M.; Viljoen, A.M. Warburgia: A comprehensive review of the botany, traditional uses and phytochemistry. J. Ethnopharmacol. 2015, 165, 260–285. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Genet. 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Ma, B.; Nussinov, R. Is allostery an intrinsic property of all dynamic proteins? Proteins Struct. Funct. Bioinform. 2004, 57, 433–443. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Amitai, G.; Shemesh, A.; Sitbon, E.; Shklar, M.; Netanely, D.; Venger, I.; Pietrokovski, S. Network Analysis of Protein Structures Identifies Functional Residues. J. Mol. Biol. 2004, 344, 1135–1146. [Google Scholar] [CrossRef]
- Atilgan, A.R.; Akan, P.; Baysal, C. Small-world communication of residues and significance for protein dynamics. Biophys. J. 2004, 86, 85–91. [Google Scholar] [CrossRef]
- Astl, L.; Verkhivker, G.M. Atomistic Modeling of the ABL Kinase Regulation by Allosteric Modulators Using Structural Perturbation Analysis and Community-Based Network Reconstruction of Allosteric Communications. J. Chem. Theory Comput. 2019, 15, 3362–3380. [Google Scholar] [CrossRef]
- Stetz, G.; Verkhivker, G.M. Computational Analysis of Residue Interaction Networks and Coevolutionary Relationships in the Hsp70 Chaperones: A Community-Hopping Model of Allosteric Regulation and Communication. PLoS Comput. Biol. 2017. [Google Scholar] [CrossRef]
- Brown, D.; Penkler, D.; Sheik Amamuddy, O.; Ross, C.; Atilgan, A.R.; Atilgan, C.; Tastan Bishop, Ö. MD-TASK: A software suite for analyzing molecular dynamics trajectories. Bioinformatics 2017. [Google Scholar] [CrossRef]
- Stetz, G.; Verkhivker, G.M. Probing Allosteric Inhibition Mechanisms of the Hsp70 Chaperone Proteins Using Molecular Dynamics Simulations and Analysis of the Residue Interaction Networks. J. Chem. Inf. Model. 2016, 56, 1490–1517. [Google Scholar] [CrossRef] [PubMed]
- Santoni, D.; Paci, P.; Paola, L.D.; Giuliani, A. Are Proteins Just Coiled Cords? Local and Global Analysis of Contact Maps Reveals the Backbone-Dependent Nature of Proteins. Curr. Protein Pept. Sci. 2016, 17, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Stetz, G.; Verkhivker, G.M. Dancing through life: Molecular dynamics simulations and network-centric modeling of allosteric mechanisms in Hsp70 and Hsp110 chaperone proteins. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Kim, B.-H.; Grishin, N. V PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Dimmic, M.W.; Rest, J.S.; Mindell, D.P.; Goldstein, R.A. rtREV: An Amino Acid Substitution Matrix for Inference of Retrovirus and Reverse Transcriptase Phylogeny. J. Mol. Evol. 2002, 55, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Hatherley, R.; Brown, D.K.; Musyoka, T.M.; Penkler, D.L.; Faya, N.; Lobb, K.A.; Tastan Bishop, Ö. SANCDB: A South African natural compound database. J. Cheminform. 2015, 7. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar] [PubMed]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kmiecik, S.; Kouza, M.; Badaczewska-Dawid, A.E.; Kloczkowski, A.; Kolinski, A. Modeling of Protein Structural Flexibility and Large-Scale Dynamics: Coarse-Grained Simulations and Elastic Network Models. Int. J. Mol. Sci. 2018, 19, 3496. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.P.; Badaczewska-Dawid, A.E.; Pikuzinska, M.; Kolinski, A.; Kmiecik, S. Modeling of disordered protein structures using monte carlo simulations and knowledge-based statistical force fields. Int. J. Mol. Sci. 2019, 20, 606. [Google Scholar] [CrossRef]
- Kurcinski, M.; Oleniecki, T.; Ciemny, M.P.; Kuriata, A.; Kolinski, A.; Kmiecik, S. CABS-flex standalone: A simulation environment for fast modeling of protein flexibility. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA: Protein complexes. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar] [CrossRef]
- Vijayabaskar, M.S.; Vishveshwara, S. Interaction energy based protein structure networks. Biophys. J. 2010. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Parekh, N. NAPS: Network analysis of protein structures. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Naganathan, V.; Garg, K.; Agarwal, Y.; Parekh, N. NAPS update: Network analysis of molecular dynamics data and protein–nucleic acid complexes. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
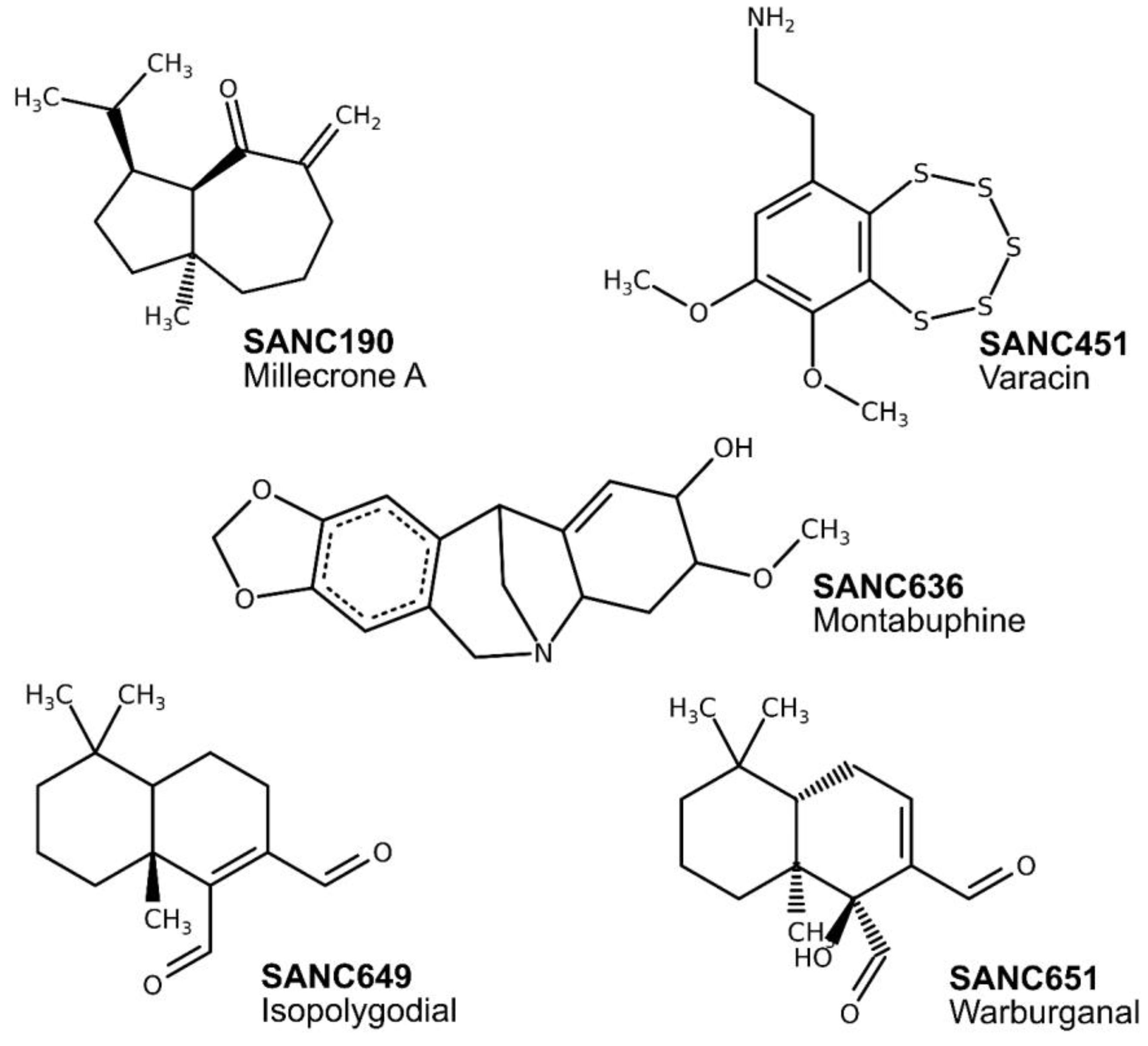
| Compound | Name | Chemical Formula | PfHsp70-x | PfHsp70-1 | ||
|---|---|---|---|---|---|---|
| Cluster Size | Average Binding Energy (kcal/mol) | Cluster Size | Average Binding Energy (kcal/mol) | |||
| SANC190 | Millecrone A | C15H24O | 75 | −7.13 | 65 | −7.03 |
| SANC451 | Varacin | C10H13NO2S5 | 45 | −8.68 | 39 | −8.73 |
| SANC636 | Montabuphine | C17H19NO4 | 83 | −8.82 | 58 | −8.26 |
| SANC649 | Isopolygodial | C15H2202 | 73 | −7.23 | 31 | −7.16 |
| SANC651 | Warburganal | C15H2203 | 68 | −7.47 | 26 | −7.70 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amusengeri, A.; Astl, L.; Lobb, K.; Verkhivker, G.M.; Tastan Bishop, Ö. Establishing Computational Approaches Towards Identifying Malarial Allosteric Modulators: A Case Study of Plasmodium falciparum Hsp70s. Int. J. Mol. Sci. 2019, 20, 5574. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225574
Amusengeri A, Astl L, Lobb K, Verkhivker GM, Tastan Bishop Ö. Establishing Computational Approaches Towards Identifying Malarial Allosteric Modulators: A Case Study of Plasmodium falciparum Hsp70s. International Journal of Molecular Sciences. 2019; 20(22):5574. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225574
Chicago/Turabian StyleAmusengeri, Arnold, Lindy Astl, Kevin Lobb, Gennady M. Verkhivker, and Özlem Tastan Bishop. 2019. "Establishing Computational Approaches Towards Identifying Malarial Allosteric Modulators: A Case Study of Plasmodium falciparum Hsp70s" International Journal of Molecular Sciences 20, no. 22: 5574. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225574