Altered Secretome and ROS Production in Olfactory Mucosa Stem Cells Derived from Friedreich’s Ataxia Patients

 ,
,
Abstract
:1. Introduction
2. Results
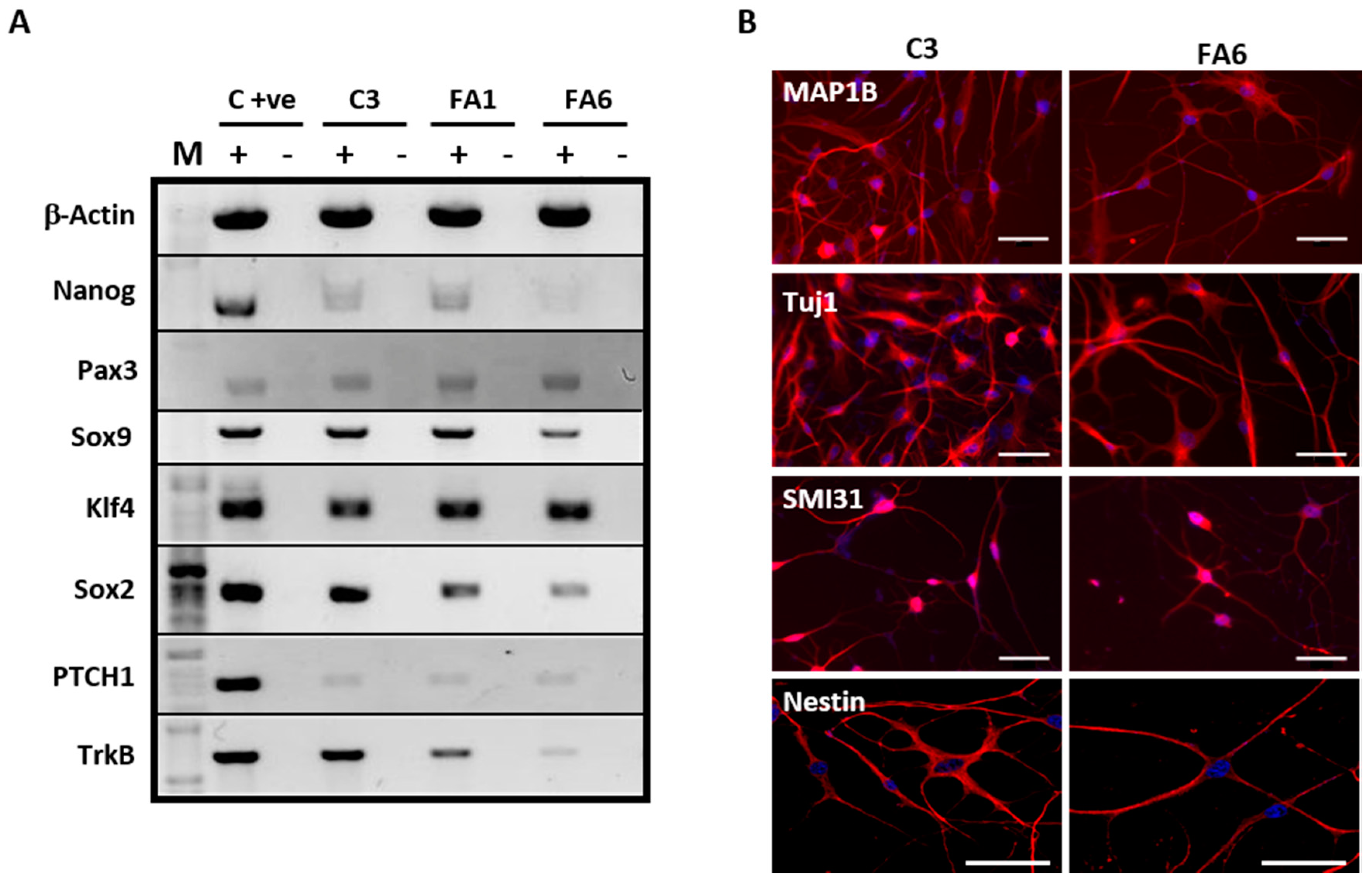
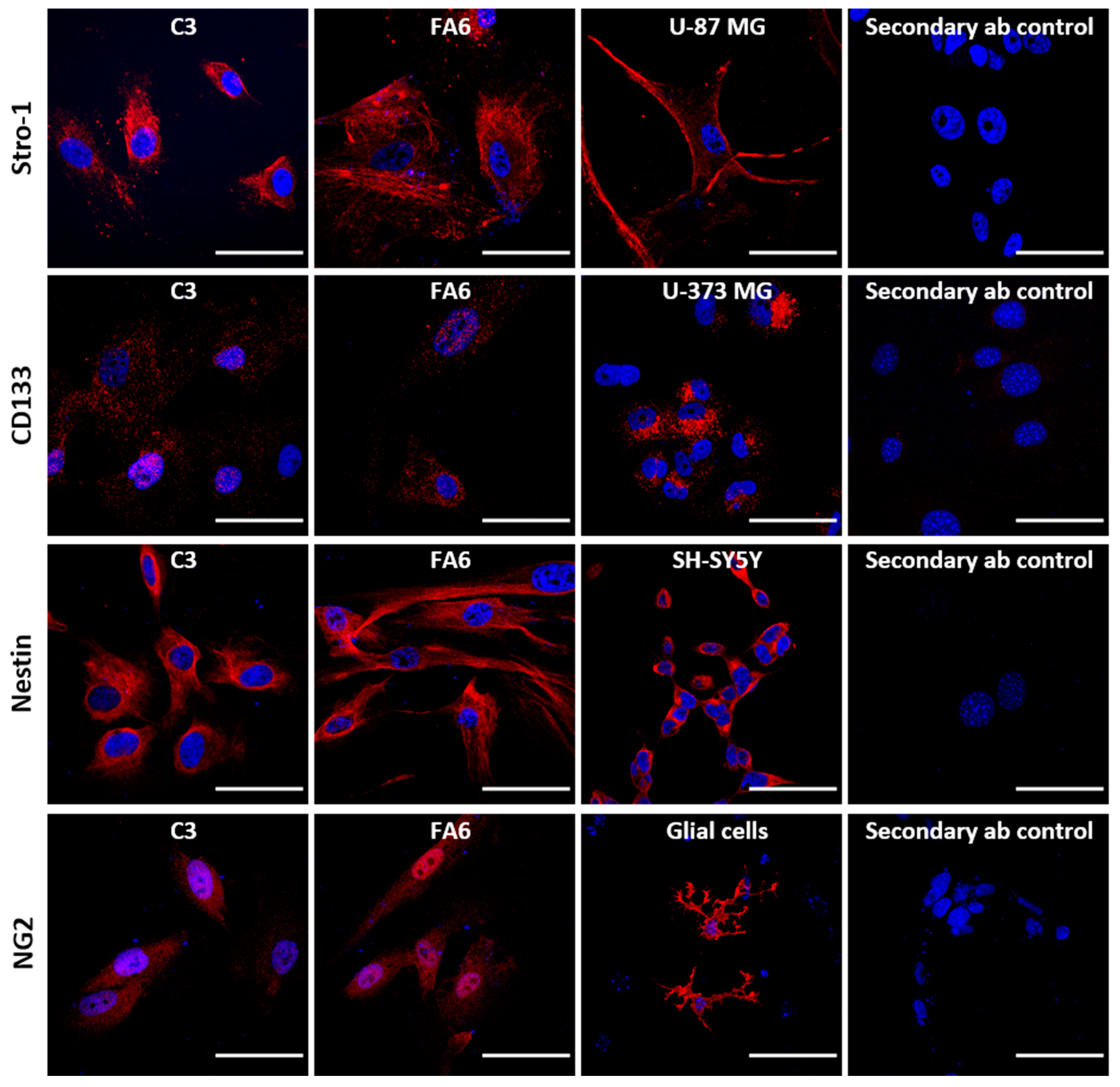
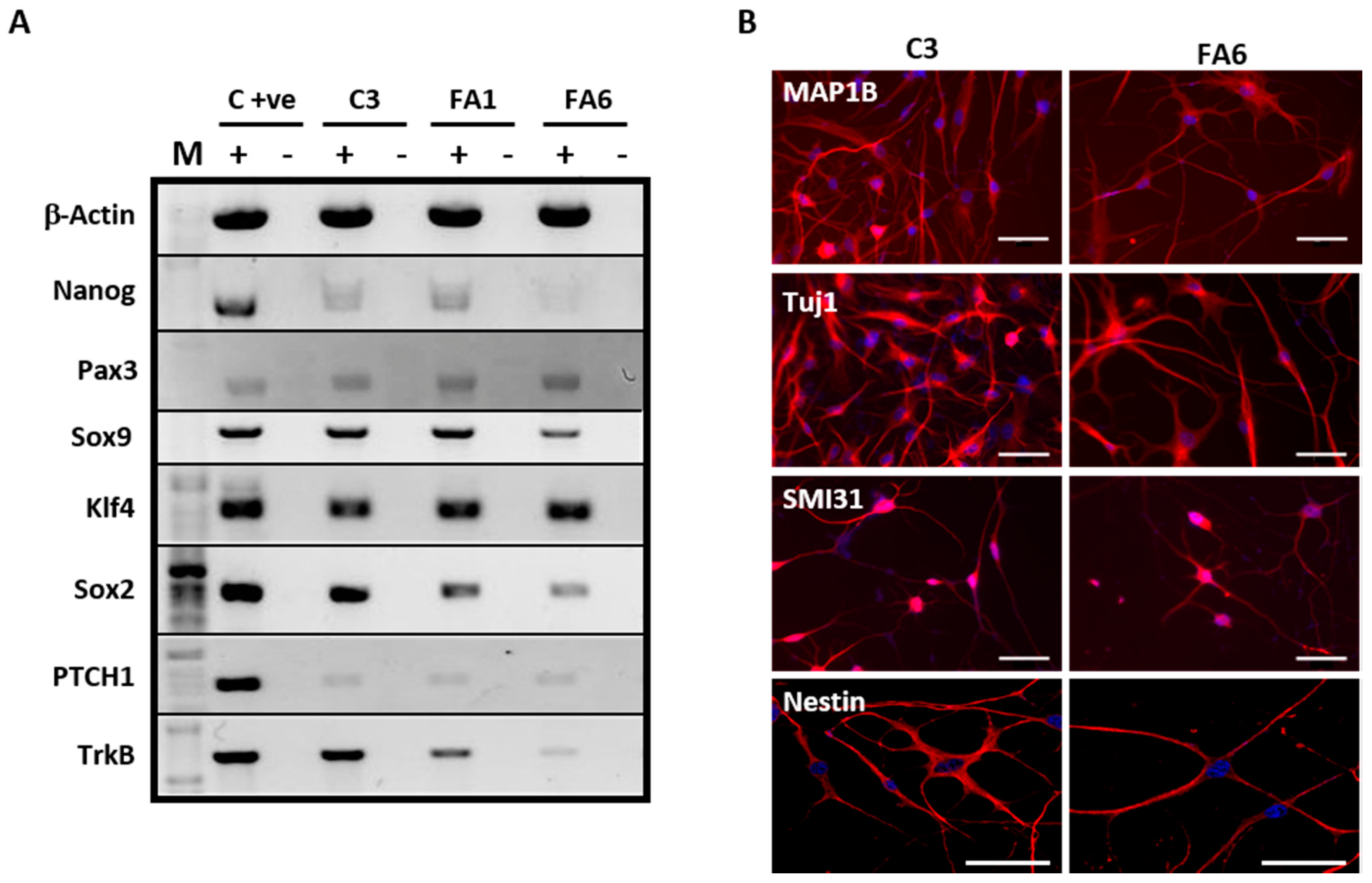
2.1. Olfactory Mucosa Stem Cells Isolated from Biopsies Exhibit a Phenotype Corresponding to Neural Crest-Derived Mesenchymal Stem Cells
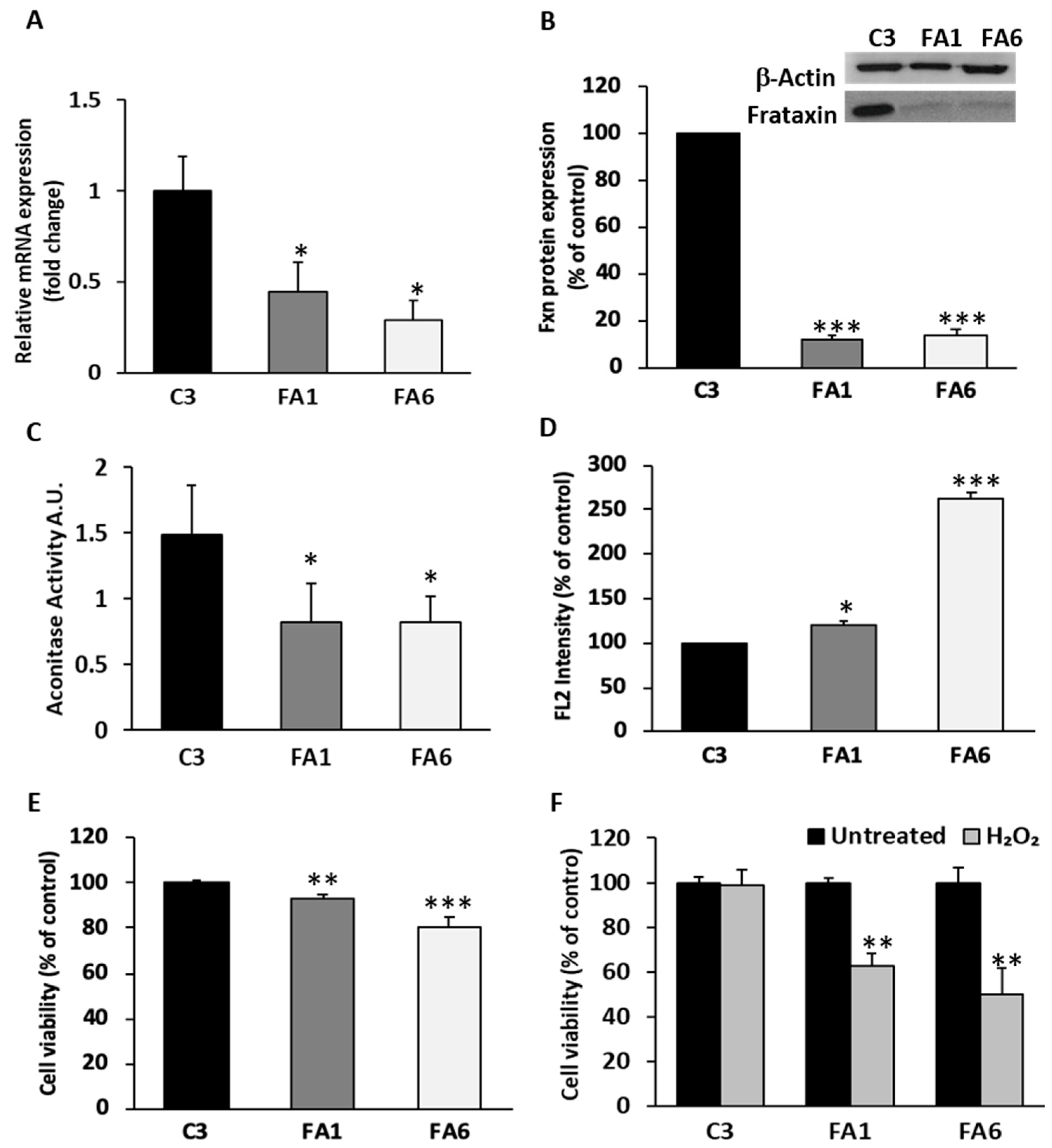
2.2. OE-MSCs from FRDA Patients Express Low Frataxin Levels, Exhibit Increased ROS Production and Decreased Cell Viability
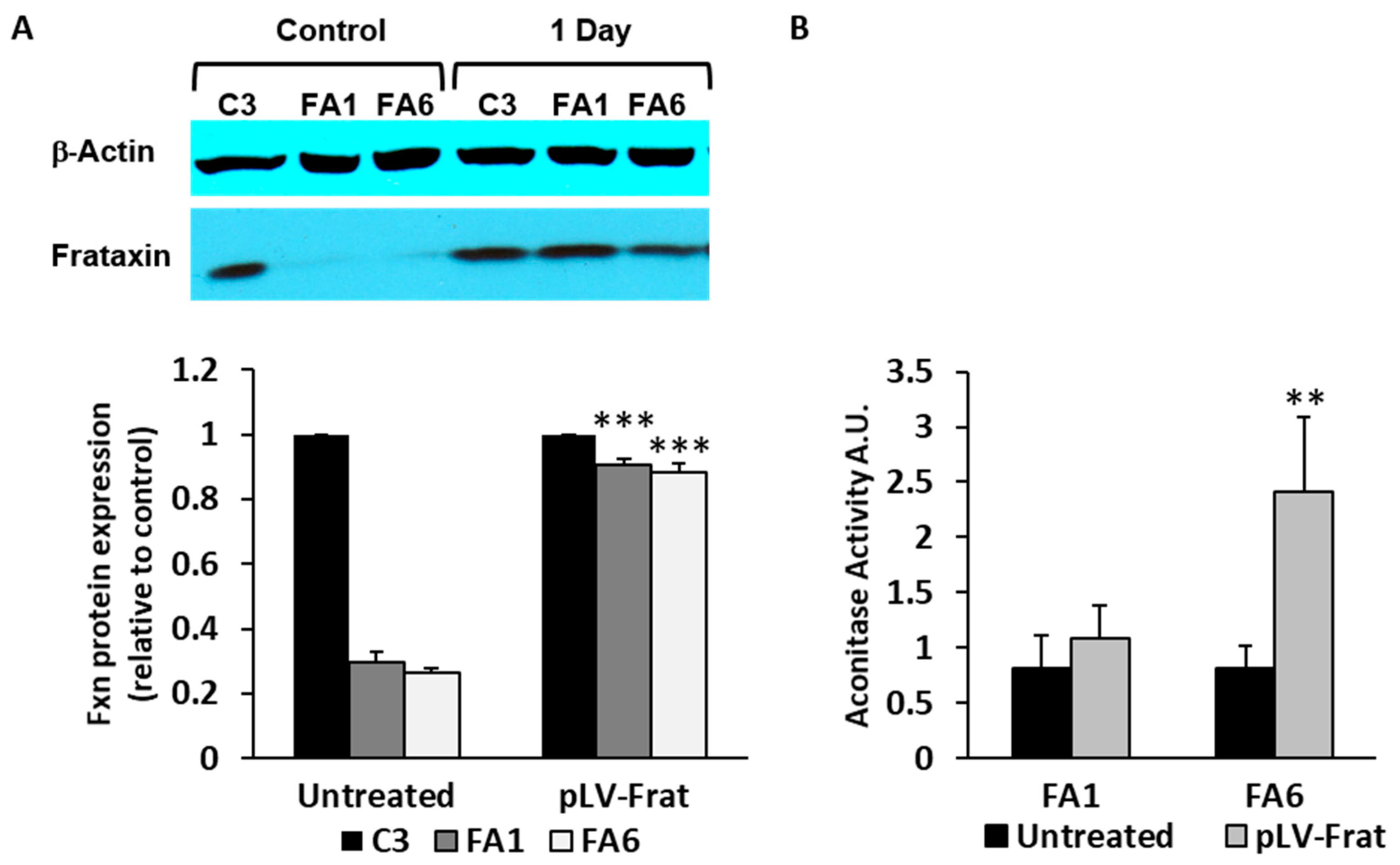
2.3. Aconitase Activity is Restored in Patient-Derived OE-MSCs Transduced with Lentivirus Encoding for Frataxin
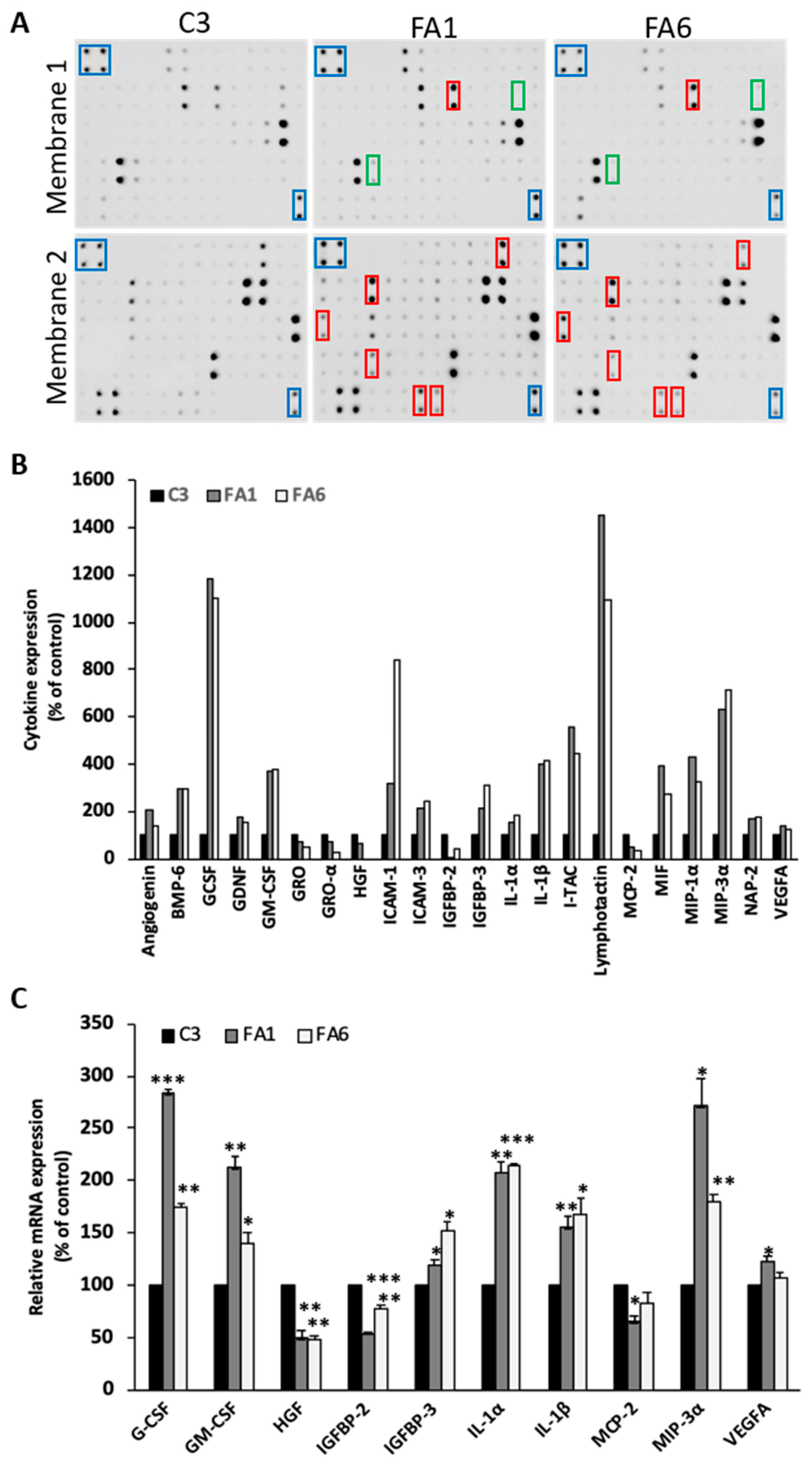
2.4. Up-regulation of Pro-Inflammatory Cytokines in Friedreich´s Ataxia Patient-Derived OE-MSCs
3. Discussion
3.1. Olfactory Stem Cells Isolated from FRDA Patients Share Features Corresponding to OE-MSCs
3.2. Phenotype of FRDA-Derived Olfactory Mucosa Cells
3.3. Cytokine Profile of FRDA-Derived OE-MSCs
4. Materials and Methods
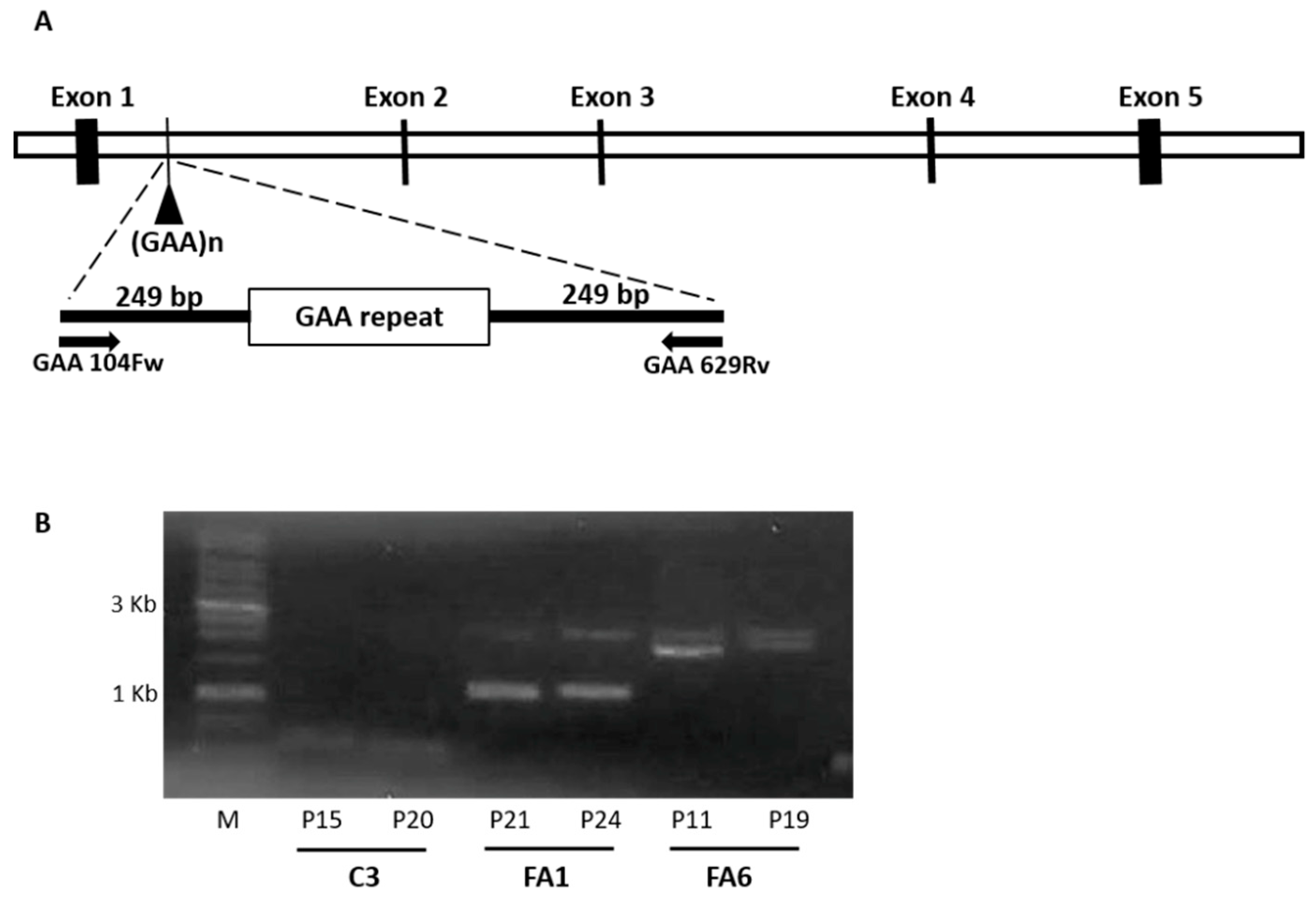
4.1. Patients
4.2. Cell Culture and Differentiation Protocol
4.3. Lentiviral Production, Titration and Cell Transduction
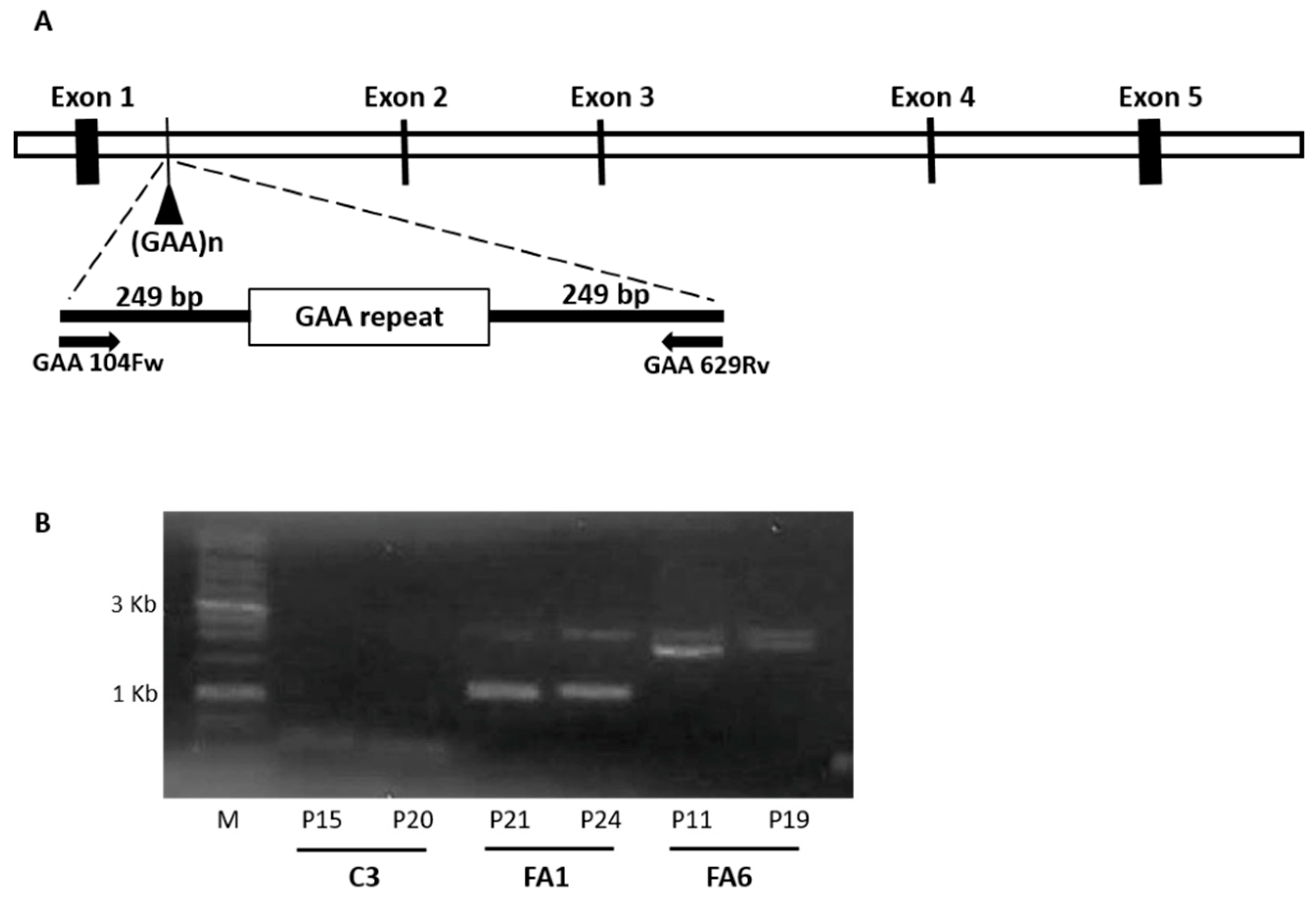
4.4. GAA Repeat Amplification
4.5. RNA Isolation, cDNA Synthesis and PCR
4.6. Quantitative PCR (qPCR)
4.7. Cell Lysis and Western Blotting
4.8. Immunocytochemistry
4.9. Reactive Oxygen Species (ROS) Generation
4.10. Aconitase Activity Measurements
4.11. Cell Viability Assays
4.12. Human Antibody-Based Protein Arrays
4.13. Data Processing and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, F.; Stork, S.; Liu, D.; Hu, K.; Herrmann, S.; Ertl, G.; Niemann, M. Cardiomyopathy of Friedreich ataxia. J. Neurochem. 2013, 126, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Mulder, H.; Igoillo-Esteve, M. Diabetes in Friedreich ataxia. J. Neurochem. 2013, 126, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Evans-Galea, M.V.; Lockhart, P.J.; Galea, C.A.; Hannan, A.J.; Delatycki, M.B. Beyond loss of frataxin: The complex molecular pathology of Friedreich ataxia. Discov. Med. 2014, 17, 25–35. [Google Scholar] [PubMed]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Yandim, C.; Natisvili, T.; Festenstein, R. Gene regulation and epigenetics in Friedreich’s ataxia. J. Neurochem. 2013, 126, 21–42. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Wells, R.D. DNA triplexes and Friedreich ataxia. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 1625–1634. [Google Scholar] [CrossRef] [Green Version]
- Gakh, O.; Bedekovics, T.; Duncan, S.F.; Smith, D.Y., IV; Berkholz, D.S.; Isaya, G. Normal and Friedreich ataxia cells express different isoforms of frataxin with complementary roles in iron-sulfur cluster assembly. J. Biol. Chem. 2010, 285, 38486–38501. [Google Scholar] [CrossRef] [Green Version]
- Vaubel, R.A.; Isaya, G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell Neurosci. 2013, 55, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Busi, M.V.; Gomez-Casati, D.F. Exploring frataxin function. IUBMB Life 2012, 64, 56–63. [Google Scholar] [CrossRef]
- Zhang, S.; Napierala, M.; Napierala, J.S. Therapeutic prospects for Friedreich’s ataxia. Trends Pharmacol. Sci. 2019, 40, 229–233. [Google Scholar] [CrossRef]
- Ahmadian-Moghadam, H.; Sadat-Shirazi, M.-S.; Zarrindast, M.-R. Therapeutic potential of stem cells for treatment of neurodegenerative diseases. Biotechnol. Lett. 2020, 42, 1073–1101. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rozwadowska, N.; Clark, A.; Fil, D.; Napierala, J.S.; Napierala, M. Excision of the expanded GAA repeats corrects cardiomyopathy phenotypes of iPSC-derived Friedreich’s ataxia cardiomyocytes. Stem Cell Res. 2019, 40, 101529. [Google Scholar] [CrossRef]
- Li, L.; Shen, X.; Liu, Z.; Norrbom, M.; Prakash, T.P.; O’Reilly, D.; Sharma, V.K.; Damha, M.J.; Watts, J.K.; Rigo, F.; et al. Activation of Frataxin Protein Expression by Antisense Oligonucleotides Targeting the Mutant Expanded Repeat. Nucleic Acid Ther. 2018, 28, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Cassina, A.; Wade-Martins, R.; Gomez-Sebastian, S.; Corona, J.C.; Lim, F.; Diaz-Nido, J. Infectious delivery and long-term persistence of transgene expression in the brain by a 135-kb iBAC-FXN genomic DNA expression vector. Gene Ther. 2011, 18, 1015–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Sebastian, S.; Gimenez-Cassina, A.; Diaz-Nido, J.; Lim, F.; Wade-Martins, R. Infectious delivery and expression of a 135 kb human FRDA genomic DNA locus complements Friedreich’s ataxia deficiency in human cells. Mol. Ther. 2007, 15, 248–254. [Google Scholar] [CrossRef]
- Perdomini, M.; Hick, A.; Puccio, H.; Pook, M.A. Animal and cellular models of Friedreich ataxia. J. Neurochem. 2013, 126, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Palomo, G.M.; Cerrato, T.; Gargini, R.; Diaz-Nido, J. Silencing of frataxin gene expression triggers p53-dependent apoptosis in human neuron-like cells. Hum. Mol. Genet. 2011, 20, 2807–2822. [Google Scholar] [CrossRef]
- Loria, F.; Diaz-Nido, J. Frataxin knockdown in human astrocytes triggers cell death and the release of factors that cause neuronal toxicity. Neurobiol. Dis. 2015, 76, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Matigian, N.A.; McCurdy, R.D.; Feron, F.; Perry, C.; Smith, H.; Filippich, C.; McLean, D.; McGrath, J.; Mackay-Sim, A.; Mowry, B.; et al. Fibroblast and lymphoblast gene expression profiles in schizophrenia: Are non-neural cells informative? PLoS ONE 2008, 3, e2412. [Google Scholar] [CrossRef] [PubMed]
- Pianese, L.; Busino, L.; De Biase, I.; De Cristofaro, T.; Lo Casale, M.S.; Giuliano, P.; Monticelli, A.; Turano, M.; Criscuolo, C.; Filla, A.; et al. Up-regulation of c-Jun N-terminal kinase pathway in Friedreich’s ataxia cells. Hum. Mol. Genet. 2002, 11, 2989–2996. [Google Scholar] [CrossRef] [PubMed]
- Haugen, A.C.; Di Prospero, N.A.; Parker, J.S.; Fannin, R.D.; Chou, J.; Meyer, J.N.; Halweg, C.; Collins, J.B.; Durr, A.; Fischbeck, K.; et al. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: Cellular model of pathology. PLoS Genet. 2010, 6, e1000812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, M.J.; Needham, K.; Frazier, A.E.; van Rooijen, J.; Leung, J.; Hough, S.; Denham, M.; Thornton, M.E.; Parish, C.L.; Nayagam, B.A.; et al. Functional characterization of Friedreich ataxia iPS-derived neuronal progenitors and their integration in the adult brain. PLoS ONE 2014, 9, e101718. [Google Scholar] [CrossRef]
- Cieslar-Pobuda, A.; Knoflach, V.; Ringh, M.V.; Stark, J.; Likus, W.; Siemianowicz, K.; Ghavami, S.; Hudecki, A.; Green, J.L.; Los, M.J. Transdifferentiation and reprogramming: Overview of the processes, their similarities and differences. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1359–1369. [Google Scholar] [CrossRef]
- Mackay-Sim, A. Concise review: Patient-derived olfactory stem cells: New models for brain diseases. Stem Cells 2012, 30, 2361–2365. [Google Scholar] [CrossRef]
- Garcia-Escudero, V.; Rosales, M.; Munoz, J.L.; Scola, E.; Medina, J.; Khalique, H.; Garaulet, G.; Rodriguez, A.; Lim, F. Patient-derived olfactory mucosa for study of the non-neuronal contribution to amyotrophic lateral sclerosis pathology. J. Cell Mol. Med. 2015, 19, 1284–1295. [Google Scholar] [CrossRef] [Green Version]
- Feron, F.; Perry, C.; McGrath, J.J.; Mackay-Sim, A. New techniques for biopsy and culture of human olfactory epithelial neurons. Arch Otolaryngol. Head Neck Surg. 1998, 124, 861–866. [Google Scholar] [CrossRef] [Green Version]
- Winstead, W.; Marshall, C.T.; Lu, C.L.; Klueber, K.M.; Roisen, F.J. Endoscopic biopsy of human olfactory epithelium as a source of progenitor cells. Am. J. Rhinol. 2005, 19, 83–90. [Google Scholar] [CrossRef]
- Leung, C.T.; Coulombe, P.A.; Reed, R.R. Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nat. Neurosci. 2007, 10, 720–726. [Google Scholar] [CrossRef]
- Murrell, W.; Feron, F.; Wetzig, A.; Cameron, N.; Splatt, K.; Bellette, B.; Bianco, J.; Perry, C.; Lee, G.; Mackay-Sim, A. Multipotent stem cells from adult olfactory mucosa. Dev. Dyn. 2005, 233, 496–515. [Google Scholar] [CrossRef] [PubMed]
- Tome, M.; Lindsay, S.L.; Riddell, J.S.; Barnett, S.C. Identification of nonepithelial multipotent cells in the embryonic olfactory mucosa. Stem Cells 2009, 27, 2196–2208. [Google Scholar] [CrossRef] [PubMed]
- Delorme, B.; Nivet, E.; Gaillard, J.; Haupl, T.; Ringe, J.; Deveze, A.; Magnan, J.; Sohier, J.; Khrestchatisky, M.; Roman, F.S.; et al. The human nose harbors a niche of olfactory ectomesenchymal stem cells displaying neurogenic and osteogenic properties. Stem Cells Dev. 2010, 19, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, Y.; Kano, S.; Ishizuka, K.; Cascella, N.G.; Ishii, S.; Talbot, C.C., Jr.; Jaffe, A.E.; Okano, H.; Pevsner, J.; Colantuoni, C.; et al. Olfactory cells via nasal biopsy reflect the developing brain in gene expression profiles: Utility and limitation of the surrogate tissues in research for brain disorders. Neurosci. Res. 2013, 77, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meirelles Lda, S.; Nardi, N.B. Methodology, biology and clinical applications of mesenchymal stem cells. Front. Biosci. Landmark Ed. 2009, 14, 4281–4298. [Google Scholar] [CrossRef]
- Marshall, C.T.; Guo, Z.; Lu, C.; Klueber, K.M.; Khalyfa, A.; Cooper, N.G.; Roisen, F.J. Human adult olfactory neuroepithelial derived progenitors retain telomerase activity and lack apoptotic activity. Brain Res. 2005, 1045, 45–56. [Google Scholar] [CrossRef]
- Lindsay, S.L.; Johnstone, S.A.; Mountford, J.C.; Sheikh, S.; Allan, D.B.; Clark, L.; Barnett, S.C. Human mesenchymal stem cells isolated from olfactory biopsies but not bone enhance CNS myelination in vitro. Glia 2013, 61, 368–382. [Google Scholar] [CrossRef]
- Veron, A.D.; Bienboire-Frosini, C.; Feron, F.; Codecasa, E.; Deveze, A.; Royer, D.; Watelet, P.; Asproni, P.; Sadelli, K.; Chabaud, C.; et al. Isolation and characterization of olfactory ecto-mesenchymal stem cells from eight mammalian genera. BMC Vet. Res. 2018, 14, 17. [Google Scholar] [CrossRef]
- Lu, W.; Duan, D.; Ackbarkhan, Z.; Lu, M.; Huang, M.L. Differentiation of human olfactory mucosa mesenchymal stem cells into photoreceptor cells in vitro. Int. J. Ophthalmol. 2017, 10, 1504–1509. [Google Scholar] [CrossRef]
- Muller, J.; Ossig, C.; Greiner, J.F.; Hauser, S.; Fauser, M.; Widera, D.; Kaltschmidt, C.; Storch, A.; Kaltschmidt, B. Intrastriatal transplantation of adult human neural crest-derived stem cells improves functional outcome in parkinsonian rats. Stem Cells Transl. Med. 2015, 4, 31–43. [Google Scholar] [CrossRef]
- Lindsay, S.L.; Toft, A.; Griffin, J.; MM Emraja, A.; Barnett, S.C.; Riddell, J.S. Human olfactory mesenchymal stromal cell transplants promote remyelination and earlier improvement in gait co-ordination after spinal cord injury. Glia 2017, 65, 639–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alizadeh, R.; Bagher, Z.; Kamrava, S.K.; Falah, M.; Ghasemi Hamidabadi, H.; Eskandarian Boroujeni, M.; Mohammadi, F.; Khodaverdi, S.; Zare-Sadeghi, A.; Olya, A.; et al. Differentiation of human mesenchymal stem cells (MSC) to dopaminergic neurons: A comparison between Wharton’s Jelly and olfactory mucosa as sources of MSCs. J. Chem. Neuroanat. 2019, 96, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Bagher, Z.; Kamrava, S.K.; Alizadeh, R.; Farhadi, M.; Absalan, M.; Falah, M.; Faghihi, F.; Zare-Sadeghi, A.; Komeili, A. Differentiation of neural crest stem cells from nasal mucosa into motor neuron-like cells. J. Chem. Neuroanat. 2018, 92, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Borgmann-Winter, K.E.; Rawson, N.E.; Wang, H.Y.; Wang, H.; Macdonald, M.L.; Ozdener, M.H.; Yee, K.K.; Gomez, G.; Xu, J.; Bryant, B.; et al. Human olfactory epithelial cells generated in vitro express diverse neuronal characteristics. Neuroscience 2009, 158, 642–653. [Google Scholar] [CrossRef] [Green Version]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, S.; Higa, K.; Igarashi, T.; Takaichi, S.; Tonogi, M.; Shinozaki, N.; Shimazaki, J.; Yamane, G.Y. Characterization of mesenchymal progenitor cell populations from non-epithelial oral mucosa. Oral. Dis. 2015, 21, 361–372. [Google Scholar] [CrossRef]
- Zhang, W.; Sui, Y.; Ni, J.; Yang, T. Insights into the Nanog gene: A propeller for stemness in primitive stem cells. Int. J. Biol. Sci. 2016, 12, 1372–1381. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; McClintick, J.; Zhong, L.; Edenberg, H.J.; Yoder, M.C.; Chan, R.J. Murine embryonic stem cell differentiation is promoted by SOCS-3 and inhibited by the zinc finger transcription factor Klf4. Blood 2005, 105, 635–637. [Google Scholar] [CrossRef]
- Vidal, V.P.; Chaboissier, M.C.; Lutzkendorf, S.; Cotsarelis, G.; Mill, P.; Hui, C.C.; Ortonne, N.; Ortonne, J.P.; Schedl, A. Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr. Biol. 2005, 15, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Avilion, A.A.; Nicolis, S.K.; Pevny, L.H.; Perez, L.; Vivian, N.; Lovell-Badge, R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003, 17, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Du, J.; Lv, Y.; Lin, H.; Mao, Z.; Xu, M.; Liu, M.; Liu, Y. PAX3 inhibits beta-Tubulin-III expression and neuronal differentiation of neural stem cell. Biochem. Biophys. Res. Commun. 2017, 485, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Drnevich, J.; Akraiko, T.; Band, M.; Li, D.; Wang, F.; Matoba, R.; Tanaka, T.S. Gene expression profiling reveals the heterogeneous transcriptional activity of Oct3/4 and its possible interaction with Gli2 in mouse embryonic stem cells. Genomics 2013, 102, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delatycki, M.B.; Williamson, R.; Forrest, S.M. Friedreich ataxia: An overview. J. Med. Genet. 2000, 37, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, J.; Volbeda, A.; Carpentier, P.; Darnault, C.; Moulis, J.M.; Fontecilla-Camps, J.C. Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure 2006, 14, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Colin, F.; Martelli, A.; Clemancey, M.; Latour, J.M.; Gambarelli, S.; Zeppieri, L.; Birck, C.; Page, A.; Puccio, H.; Ollagnier de Choudens, S. Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J. Am. Chem. Soc. 2013, 135, 733–740. [Google Scholar] [CrossRef]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef]
- Gomes, C.M.; Santos, R. Neurodegeneration in Friedreich’s ataxia: From defective frataxin to oxidative stress. Oxid. Med. Cell Longev. 2013, 2013, 487534. [Google Scholar] [CrossRef]
- Rufini, A.; Cavallo, F.; Condo, I.; Fortuni, S.; De Martino, G.; Incani, O.; Di Venere, A.; Benini, M.; Massaro, D.S.; Arcuri, G.; et al. Highly specific ubiquitin-competing molecules effectively promote frataxin accumulation and partially rescue the aconitase defect in Friedreich ataxia cells. Neurobiol. Dis. 2015, 75, 91–99. [Google Scholar] [CrossRef]
- Goncalves, S.; Paupe, V.; Dassa, E.P.; Rustin, P. Deferiprone targets aconitase: Implication for Friedreich’s ataxia treatment. BMC Neurol. 2008, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Bulteau, A.L.; O’Neill, H.A.; Kennedy, M.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 2004, 305, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, R.; Kozlov, S.; Matigian, N.; Wali, G.; Gatei, M.; Sutharsan, R.; Bellette, B.; Wraith-Kijas, A.; Cochrane, J.; Coulthard, M.; et al. A patient-derived olfactory stem cell disease model for ataxia-telangiectasia. Hum. Mol. Genet. 2013, 22, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Chagastelles, P.C.; Nardi, N.B.; Camassola, M. Biology and applications of mesenchymal stem cells. Sci. Prog. 2010, 93, 113–127. [Google Scholar] [CrossRef]
- Corti, S.; Nizzardo, M.; Nardini, M.; Donadoni, C.; Locatelli, F.; Papadimitriou, D.; Salani, S.; Del Bo, R.; Ghezzi, S.; Strazzer, S.; et al. Isolation and characterization of murine neural stem/progenitor cells based on Prominin-1 expression. Exp. Neurol. 2007, 205, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Wiese, C.; Rolletschek, A.; Kania, G.; Blyszczuk, P.; Tarasov, K.V.; Tarasova, Y.; Wersto, R.P.; Boheler, K.R.; Wobus, A.M. Nestin expression-a property of multi-lineage progenitor cells? Cell Mol. Life Sci. 2004, 61, 2510–2522. [Google Scholar] [CrossRef]
- Belachew, S.; Chittajallu, R.; Aguirre, A.A.; Yuan, X.; Kirby, M.; Anderson, S.; Gallo, V. Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J. Cell Biol. 2003, 161, 169–186. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.A.; Chittajallu, R.; Belachew, S.; Gallo, V. NG2-expressing cells in the subventricular zone are type C-like cells and contribute to interneuron generation in the postnatal hippocampus. J. Cell Biol. 2004, 165, 575–589. [Google Scholar] [CrossRef]
- Valny, M.; Honsa, P.; Kriska, J.; Anderova, M. Multipotency and therapeutic potential of NG2 cells. Biochem. Pharmacol. 2017, 141, 42–55. [Google Scholar] [CrossRef]
- Dennis, J.E.; Carbillet, J.P.; Caplan, A.I.; Charbord, P. The STRO-1+ marrow cell population is multipotential. Cells Tissues Organs 2002, 170, 73–82. [Google Scholar] [CrossRef]
- Gronthos, S.; Graves, S.E.; Ohta, S.; Simmons, P.J. The STRO-1+ fraction of adult human bone marrow contains the osteogenic precursors. Blood 1994, 84, 4164–4173. [Google Scholar] [CrossRef] [Green Version]
- Meirelles Lda, S.; Nardi, N.B. Murine marrow-derived mesenchymal stem cell: Isolation, in vitro expansion, and characterization. Br. J. Haematol. 2003, 123, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Goldring, K.; Jones, G.E.; Thiagarajah, R.; Watt, D.J. The effect of galectin-1 on the differentiation of fibroblasts and myoblasts in vitro. J. Cell Sci. 2002, 115, 355–366. [Google Scholar] [PubMed]
- Panetti, T.S. Tyrosine phosphorylation of paxillin, FAK, and p130CAS: Effects on cell spreading and migration. Front. Biosci. 2002, 7, 143–150. [Google Scholar]
- Bon, C.; Luffarelli, R.; Russo, R.; Fortuni, S.; Pierattini, B.; Santulli, C.; Fimiani, C.; Persichetti, F.; Cotella, D.; Mallamaci, A.; et al. SINEUP non-coding RNAs rescue defective frataxin expression and activity in a cellular model of Friedreich’s Ataxia. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, E.C.; Santani, A.B.; Perlman, S.L.; Farmer, J.M.; Stolle, C.A.; Marusich, M.F.; Lynch, D.R. A rapid, noninvasive immunoassay for frataxin: Utility in assessment of Friedreich ataxia. Mol. Genet. Metab. 2010, 101, 238–245. [Google Scholar] [CrossRef] [Green Version]
- Lushchak, O.V.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox. Rep. 2014, 19, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, M.; Kudin, A.P.; Kunz, W.S. Mitochondrial dysfunction in neurodegenerative disorders. Biochem. Soc. Trans. 2007, 35, 1228–1231. [Google Scholar] [CrossRef]
- Matigian, N.; Abrahamsen, G.; Sutharsan, R.; Cook, A.L.; Vitale, A.M.; Nouwens, A.; Bellette, B.; An, J.; Anderson, M.; Beckhouse, A.G.; et al. Disease-specific, neurosphere-derived cells as models for brain disorders. Dis. Model Mech. 2010, 3, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Schoenfeld, R.; Shan, Y.; Tsai, H.J.; Hammock, B.; Cortopassi, G. Frataxin deficiency induces Schwann cell inflammation and death. Biochim. Biophys. Acta 2009, 1792, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Spulber, S.; Schultzberg, M. Connection between inflammatory processes and transmittor function-Modulatory effects of interleukin-1. Prog. Neurobiol. 2010, 90, 256–262. [Google Scholar] [CrossRef]
- Das, M.; Tang, X.; Han, J.Y.; Mayilsamy, K.; Foran, E.; Biswal, M.R.; Tzekov, R.; Mohapatra, S.S.; Mohapatra, S. CCL20-CCR6 axis modulated traumatic brain injury-induced visual pathologies. J. Neuroinflammation 2019, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Leonardo, C.C.; Musso, J.; Das, M.; Rowe, D.D.; Collier, L.A.; Mohapatra, S.; Pennypacker, K.R. CCL20 Is Associated with Neurodegeneration Following Experimental Traumatic Brain Injury and Promotes Cellular Toxicity In Vitro. Transl. Stroke Res. 2012, 3, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Noma, S.; Ohya-Shimada, W.; Kanai, M.; Ueda, K.; Nakamura, T.; Funakoshi, H. Overexpression of HGF attenuates the degeneration of Purkinje cells and Bergmann glia in a knockin mouse model of spinocerebellar ataxia type 7. Neurosci. Res. 2012, 73, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef] [Green Version]
- Perez-Luz, S.; Gimenez-Cassina, A.; Fernandez-Frias, I.; Wade-Martins, R.; Diaz-Nido, J. Delivery of the 135 kb human frataxin genomic DNA locus gives rise to different frataxin isoforms. Genomics 2015, 106, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacary, E.; Legros, H.; Valable, S.; Duchatelle, P.; Lecocq, M.; Petit, E.; Nicole, O.; Bernaudin, M. Synergistic effects of CoCl(2) and ROCK inhibition on mesenchymal stem cell differentiation into neuron-like cells. J. Cell Sci. 2006, 119, 2667–2678. [Google Scholar] [CrossRef] [Green Version]
- Fleming, J.; Spinoulas, A.; Zheng, M.; Cunningham, S.C.; Ginn, S.L.; McQuilty, R.C.; Rowe, P.B.; Alexander, I.E. Partial correction of sensitivity to oxidant stress in Friedreich ataxia patient fibroblasts by frataxin-encoding adeno-associated virus and lentivirus vectors. Hum. Gene Ther. 2005, 16, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Follenzi, A.; Naldini, L. HIV-based vectors. Preparation and use. Methods Mol. Med. 2002, 69, 259–274. [Google Scholar]
- Valensi-Kurtz, M.; Lefler, S.; Cohen, M.A.; Aharonowiz, M.; Cohen-Kupiec, R.; Sheinin, A.; Ashery, U.; Reubinoff, B.; Weil, M. Enriched population of PNS neurons derived from human embryonic stem cells as a platform for studying peripheral neuropathies. PLoS ONE 2010, 5, e9290. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Locke, M.; Webb, R.D.; Roberts, J.T.; Langley, M.; Thomas, D.W.; Archer, C.W.; Stephens, P. A multipotent neural crest-derived progenitor cell population is resident within the oral mucosa lamina propria. Stem Cells Dev. 2010, 19, 819–830. [Google Scholar] [CrossRef]
- Pedrotti, B.; Ulloa, L.; Avila, J.; Islam, K. Characterization of microtubule-associated protein MAP1B: Phosphorylation state, light chains, and binding to microtubules. Biochem. 1996, 35, 3016–3023. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Hasko, G.; Hawkins, B.J.; Madesh, M.; Pacher, P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat. Protoc. 2007, 2, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Gil, E.; Esteban, M. Role of mitochondria in apoptosis induced by the 2-5A system and mechanisms involved. Apoptosis 2006, 11, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Barger, S.W.; Begley, J.G.; Mark, R.J. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995, 46, 187–216. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | Accession No. |
|---|---|---|---|
| FXN | TGGAATGTCAAAAAGCAGAGTG | CCACTCCCAAAGGAGACATC | NM_000144 |
| HGF | TCGGGGTAAAGACCTACAGGA | AATGGGGAGAGTTATCGAGGT | NM_000601.4 |
| IL1α | ATCAGTACCTCACGGCTGCT | TGGGTATCTCAGGCATCTCC | NM_000575.3 |
| MCP2 | CCGAGGAGCAGAGAGGTTGAGAAC | CTTGGGACATTGGATGTTGGTGATT | NM_005623.2 |
| MIF | ACCGCTCCTACAGCAAGC | CGCGTTCATGTCGTAATAGTTG | NM_002415.1 |
| IL1β | AAACAGATGAAGTGCTCCTTCCAGG | CATGGCCACAACAACTGACG | NM_000576.2 |
| BMP-6 | AACCTGGTGGAGTACGACAAG | TCACCCTCAGGAATCTGGGAT | NM_001718.4 |
| IGFBP2 | GGTATGAAGGAGCTGGCCGTGTTC | CGCTGCCCGTTCAGAGACATCTTG | NM_000597.2 |
| IGFBP3 | GCCAGGAAATGCTAGTGAGTCG | GGCAGGGACCATATTCTGTCT | NM_001013398 |
| MIP-3α | ACATCAATGCTATCATCTTTCACAC | CCAACCCCAGCAAGGTTCTT | NM_004591.2 |
| VEGFA | CTCACCAAGGCCAGCACATA | CCACAGGGGAACGCTCCAG | NM_001171624 |
| GM-CSF | AGAGACACTGCTGCTGAGATG | CCAGCAGTCAAAGGGGATGA | NM_000758.3 |
| G-CSF | CAGAGCCCCATGAAGCTGAT | GGAAAAGGCCGCTATGGAGT | NM_000759.3 |
| β-ACTIN | AACTCCATCATGAAGTGTGACG | GATCCACATCTGCTGGAAGG | NM_001101.3 |
| Up-Regulated Cytokines | Name |
| Angiogenesis/Proliferation | ANG, ECGF, KITLG (SCF), TGFB1, VEGFA |
| Iron regulatory proteins | BMP6 |
| Immunity/Inflammation | ENA-78, ICAM1, ICAM3, IL1α, IL1β, IL6, I-TAC, Lymphotactin (XCL1), MIF, MIP1α (CCL3), MIP3α, NAP-2, SDF (CXCL12), TARC (CCL17), TNF |
| Neurotropic factors | BDNF, GDNF |
| Growth factors | G-CSF, GM-CSF, IGF1 |
| Growth factor binding proteins | IGFBP3 |
| Down-Regulated Cytokines | Name |
| Angiogenesis/Proliferation | PIGF |
| Immunity/Inflammation | GRO- α (CXCL1), HCC4 (CCL16), MCP2 (CCL8), NTF4 |
| Growth factors | HGF, FGF |
| Growth factor binding proteins | IGFBP2 |
| Gene | Primers (5′-3′) | Tª Annealing | Extension Time |
|---|---|---|---|
| KFL4 (NM_004235) | Fw: ACCCGGGGCCCAATTACCCA Rv: AAGGCGAGGTGGTCCGACCT | 65 °C | 30″ |
| NANOG (NM_001297698) | Fw: TGATTTGTGGGCCTGAAGAA Rv: GCATGCAGGACTGCAGAGAT | 62 °C | 45″ |
| PAX3 (NM_181459) | Fw: AGCACCCCAATCAGATGAAG Rv: TGTCTGGGTTGGAAGGAATC | 61 °C | 30″ |
| PTCH1 (NM_001083602) | Fw: CGCACAGAACTCCACTCAAA Rv: GGGCCAGAAGAAAAACATCA | 61 °C | 20″ |
| SOX2 (NM_003106) | Fw: ATGTATCTCCCCGGCGCCGA Rv: TCGGCATCGCGGTTTTTGCG | 65 °C | 30″ |
| SOX9 (NM_000346) | Fw: CCGACGAGCAGGAGAAGGGCCTG Rv: TCGCGGAAGTCGATAGGGGGC | 65 °C | 45″ |
| TRKB (NM_001291937) | Fw: ACCCCCATTCGCATCTAAC Rv: CAGAAATGCTTTATGAGCCACA | 60 °C | 45″ |
| β-ACTIN (NM_001101) | Fw: CCACACTGTGCCCATCTACGAGGGGT Rv: AGGGCAGTGATCTCCTTCTGCATCCT | 60 °C | 30″ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Luz, S.; Loria, F.; Katsu-Jiménez, Y.; Oberdoerfer, D.; Yang, O.-L.; Lim, F.; Muñoz-Blanco, J.L.; Díaz-Nido, J. Altered Secretome and ROS Production in Olfactory Mucosa Stem Cells Derived from Friedreich’s Ataxia Patients. Int. J. Mol. Sci. 2020, 21, 6662. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186662
Pérez-Luz S, Loria F, Katsu-Jiménez Y, Oberdoerfer D, Yang O-L, Lim F, Muñoz-Blanco JL, Díaz-Nido J. Altered Secretome and ROS Production in Olfactory Mucosa Stem Cells Derived from Friedreich’s Ataxia Patients. International Journal of Molecular Sciences. 2020; 21(18):6662. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186662
Chicago/Turabian StylePérez-Luz, Sara, Frida Loria, Yurika Katsu-Jiménez, Daniel Oberdoerfer, Oscar-Li Yang, Filip Lim, José Luis Muñoz-Blanco, and Javier Díaz-Nido. 2020. "Altered Secretome and ROS Production in Olfactory Mucosa Stem Cells Derived from Friedreich’s Ataxia Patients" International Journal of Molecular Sciences 21, no. 18: 6662. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186662