SAAMBE-3D: Predicting Effect of Mutations on Protein–Protein Interactions

 , ,
, , 
Abstract
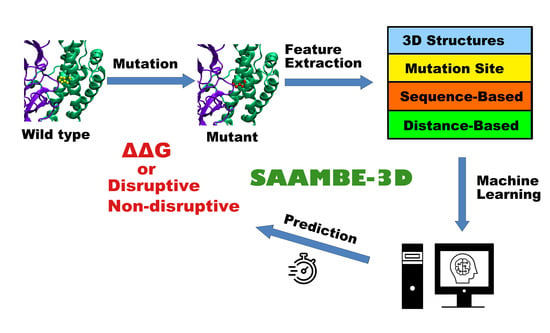
:
1. Introduction
2. Results and Discussion
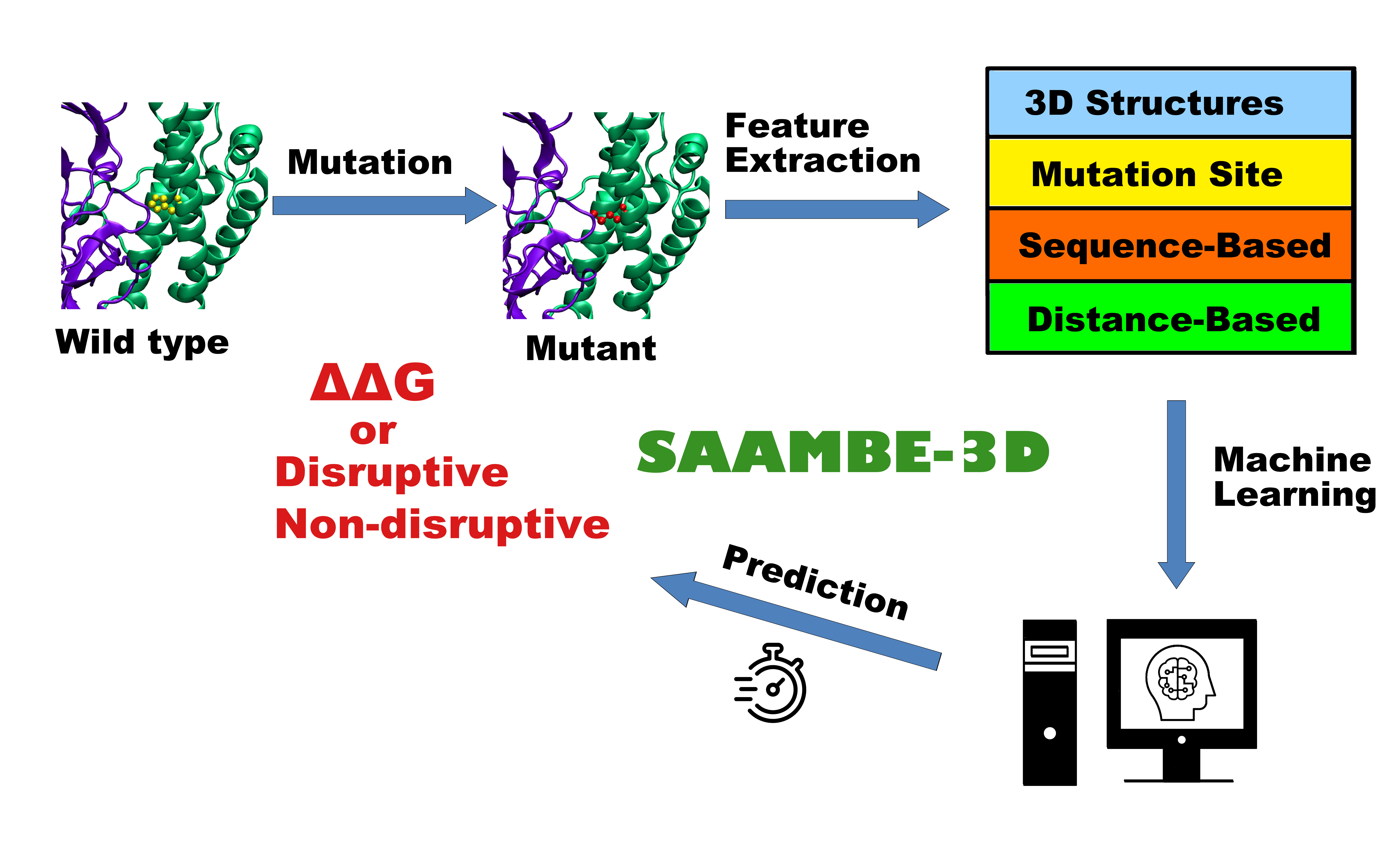
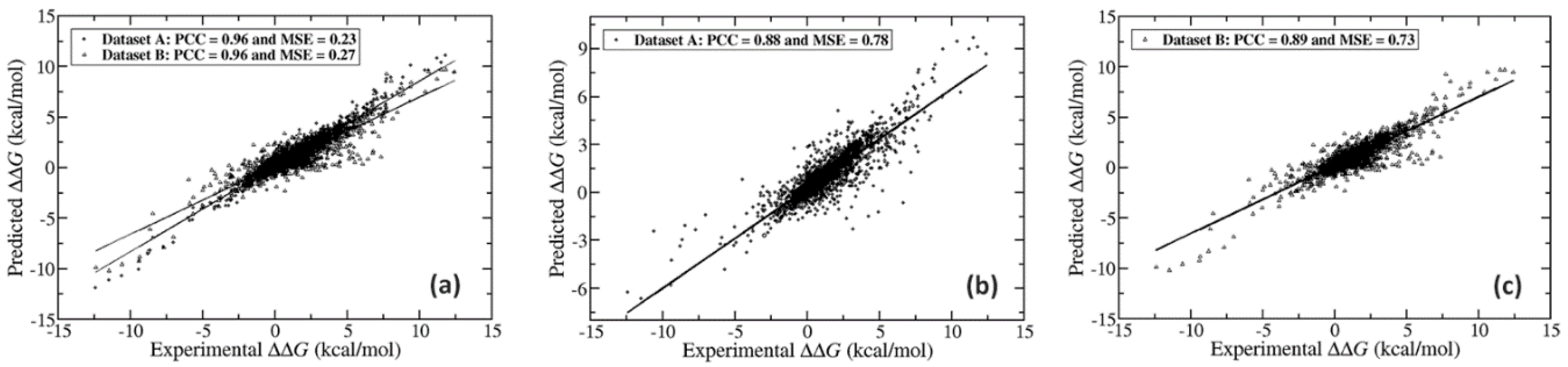
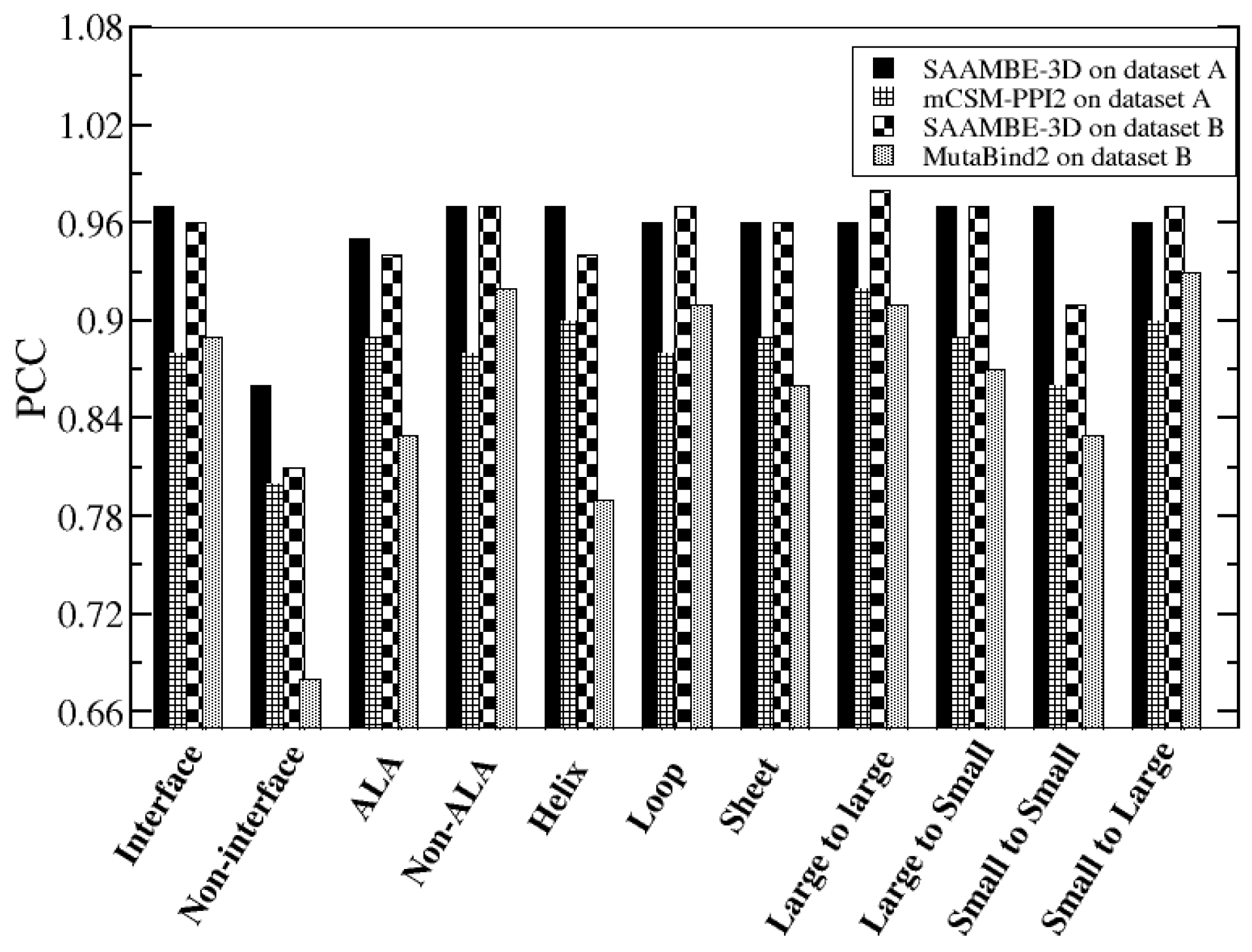
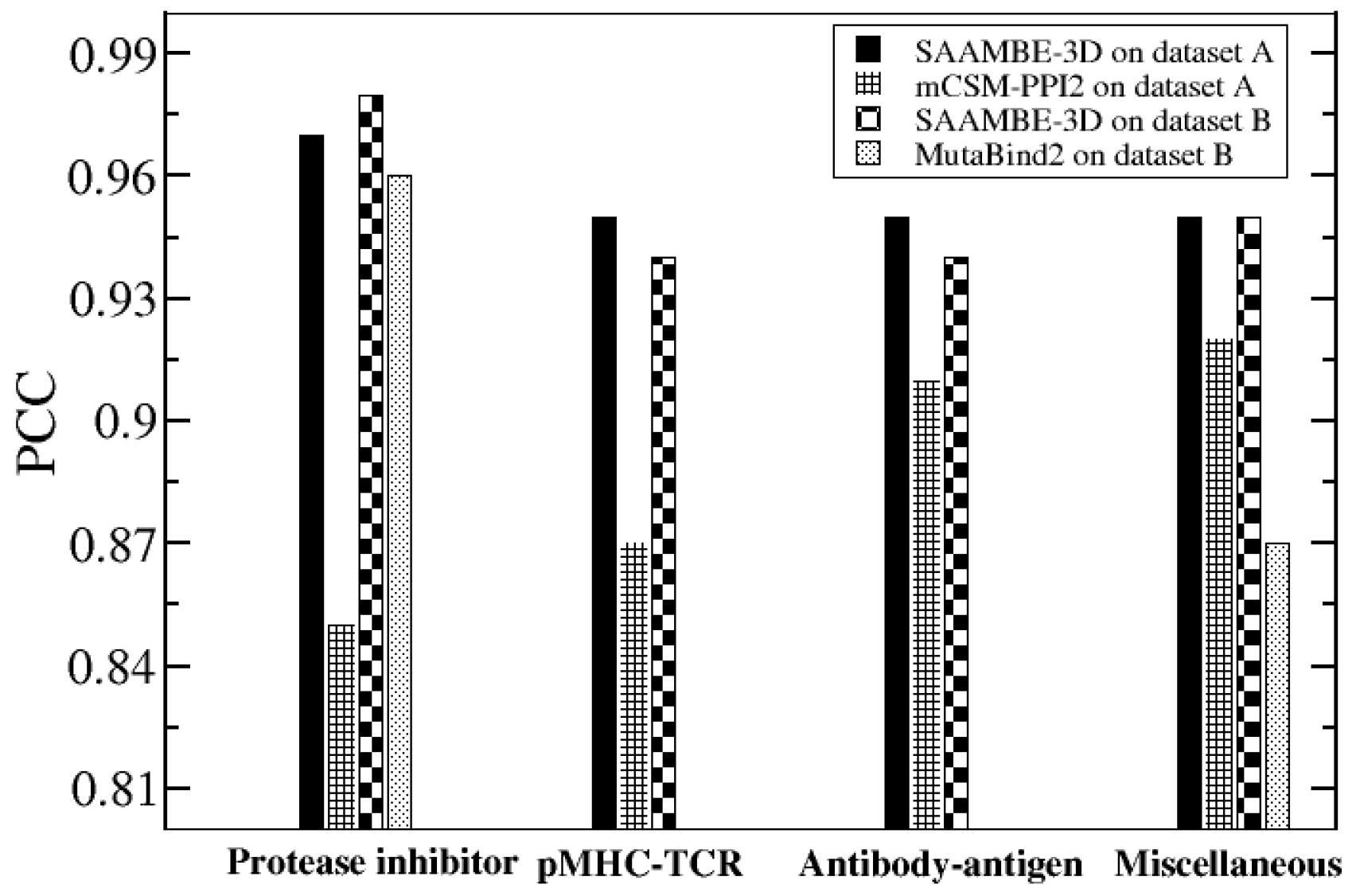
2.1. Further Performance Assessment in Comparison with Existing Methods
2.1.1. Comparison of Methods that were Trained on the Same Dataset (SKEMPI v2.0)
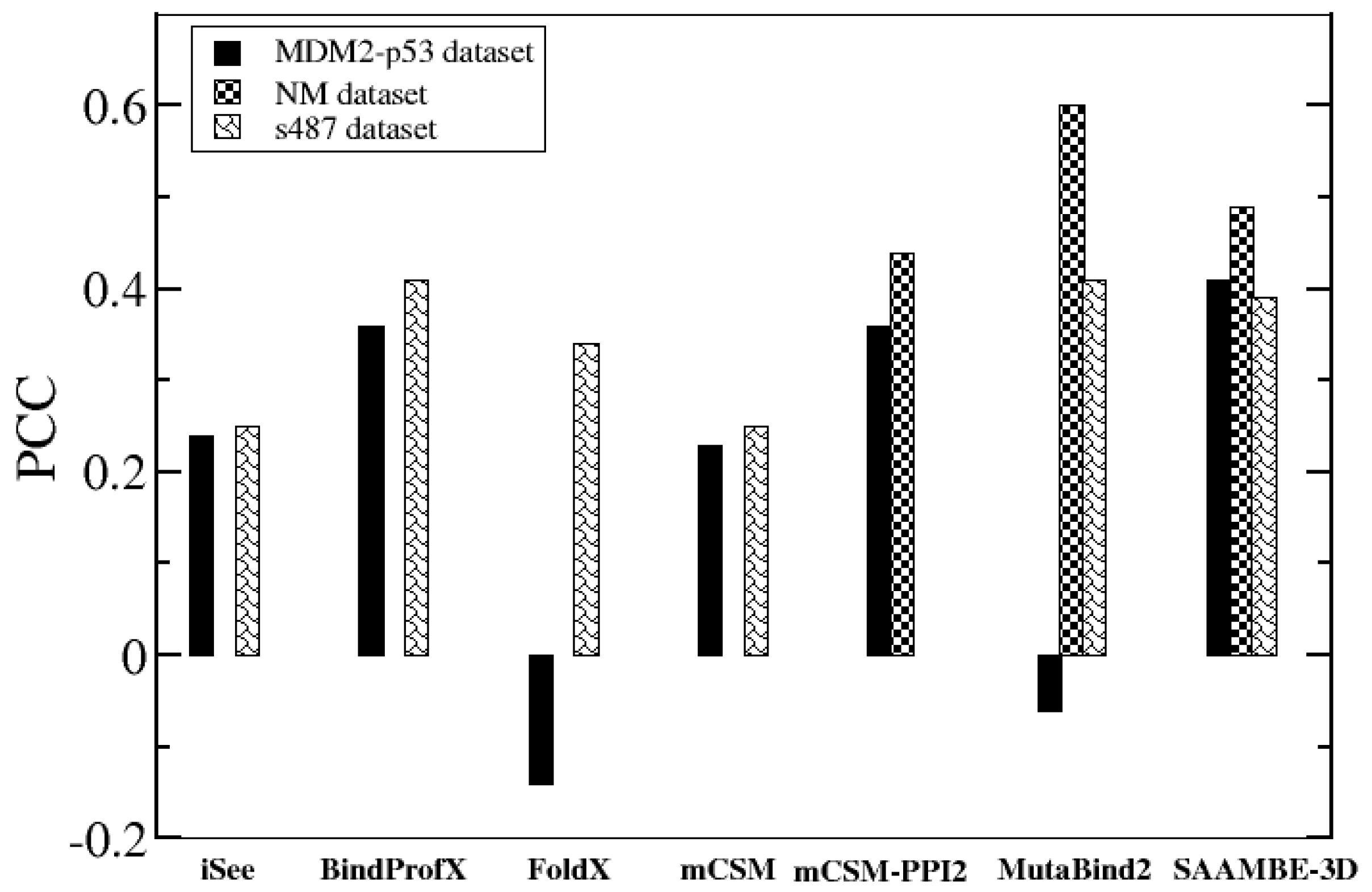
2.1.2. Performance Comparison on Blind Tests on Set of Data not Used in the Training
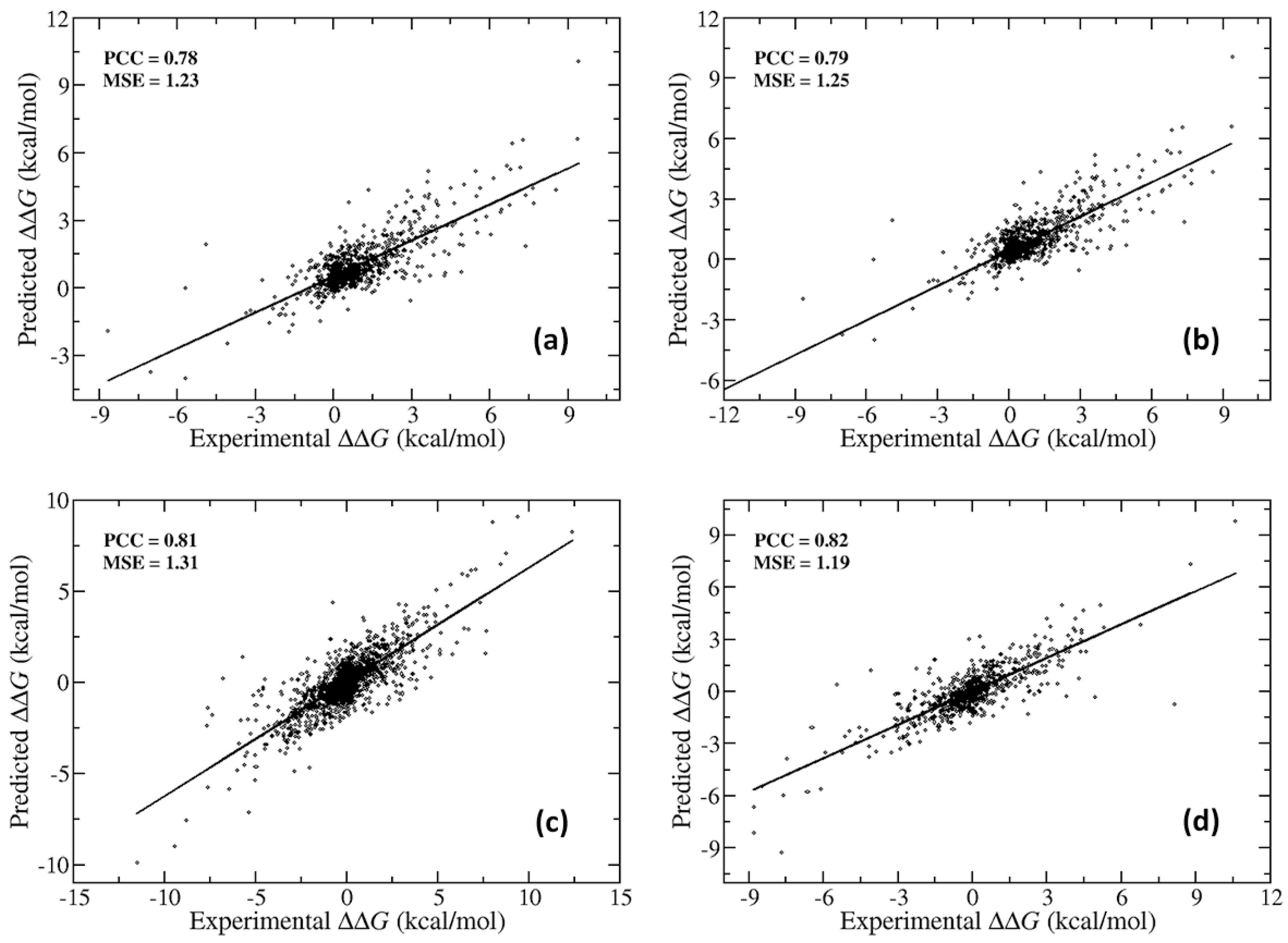
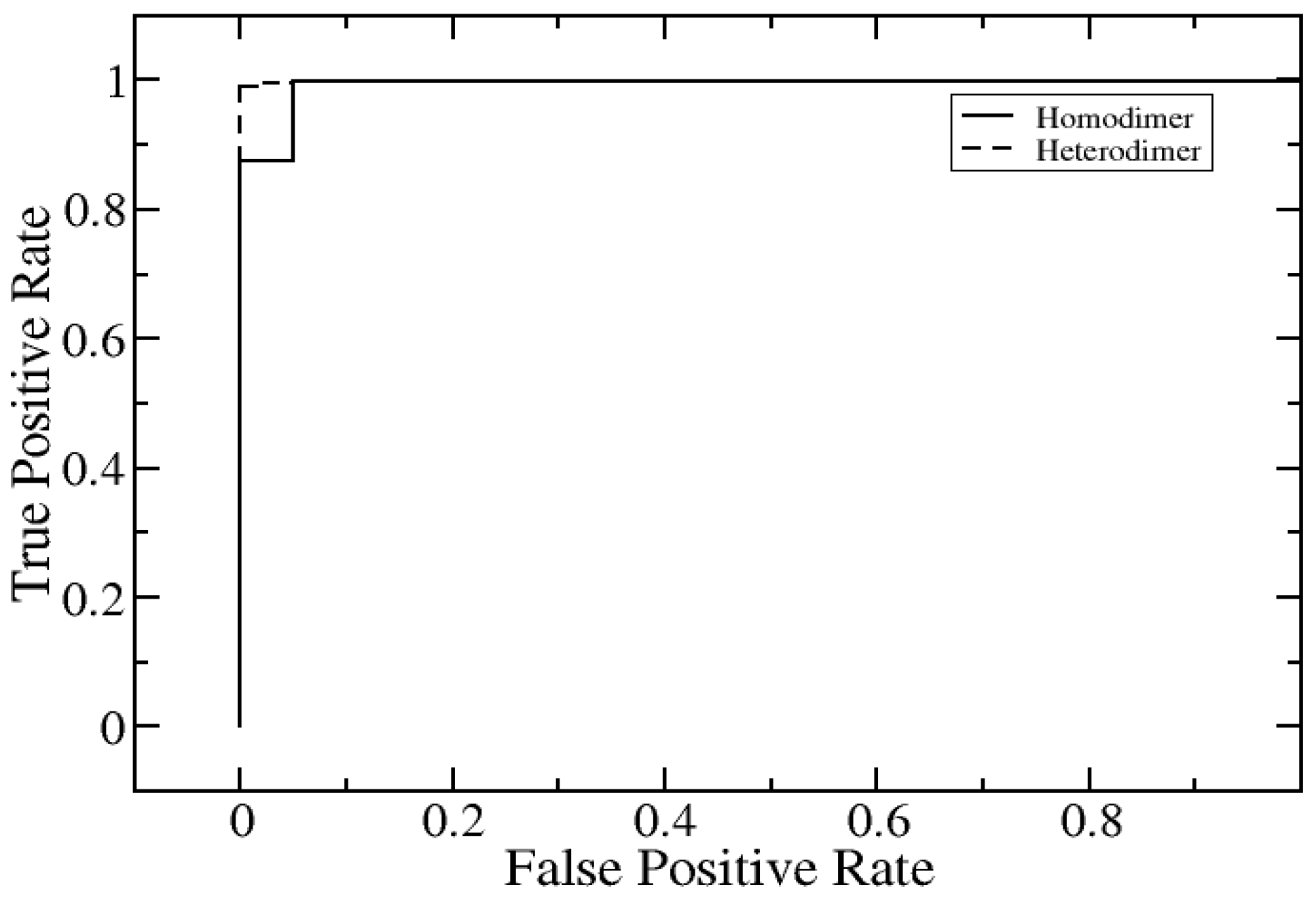
2.2. Further Development of SAAME-3D to Identify Disruptive and Non-Disruptive Mutations both in Homo- and Hetero-Dimeric Protein Complexes (Cornell University Dataset)
2.3. Time of Calculations
3. Web Server
4. Methods
4.1. Dataset Creation
4.2. Model Development
4.3. Features
4.3.1. Features Related to 3D Structures of the Protein–Protein Complex
- The temperature at which the crystal structure is obtained for each complex. This feature accounts for the effect of temperature on the structure as structures obtained at room temperature are typically more flexible (larger atomic B-factors) than structures crystalized at cryogenic temperature.
- Resolution of the PDB structures of the complex. The feature account for the structural quality of the corresponding protein–protein complex.
4.3.2. Features Related to Mutation Site
- Net volume: The feature represents the change in the molar volume of an amino acid due to mutation. For example, if in a protein–protein complex, a given residue in the wild type is mutated from arginine (R) to alanine (A), then volume change is calculated as molar volume (A)–molar volume (R).
- Net hydrophobicity: The feature accounts for the change in hydrophobic index (HI) going from wild type to mutant. For example, if in a wild type complex, alanine (A) is mutated to lysine (K) in the mutant, net hydrophobicity will be estimated by HI(K)–HI(A). We considered Moon’s hydrophobic index [60] in this study.
- Mutation type: We used the label encoding method for this feature. We labeled each different type of mutation. For example, alanine to lysine is labeled 1, lysine to glycine is labeled 2, glutamic acid to arginine is 3 and so on. In this way, there can be a possibility of 380 different types of labels.
- Net flexibility: The feature estimates flexibility through the presence of a number of rotamers for each residue. Net flexibility represents a change in the number of rotamers going from wild type to mutant residue (see [61] for more details).
- Chemical property: The label encoding method is applied to seven different chemical properties associated to amino acid residues both for wild type as well as mutant amino acid residue: (i) Aliphatic (A, G, I, L, P, V) (ii) aromatic (F, W, Y) (iii) sulfur (C, M) (iv) hydroxyl (S, T) (v) basic (R, H, K) (vi) acidic (D, E) and (vii) amide (N, Q). Thus, mutations are divided into 49 different categories corresponds to change from wild type to mutant residue through the use of this feature.
- Size: Amino acids are grouped into three classes according to their sizes: small (G, A, S, C, D, P, N, T), medium (Q, E, H, V) and large (R, I, L, K, M, F, W, Y). The label encoding method is used to create nine different labels representing small to medium, small to small, small to large, medium to medium, medium to large, medium to small, large to small, large to medium and large to a large change in the size of amino acid due to mutation.
- Polarity: Two polarity classes: polar (R, N, D, Q, E, H, K, S, T, Y) and nonpolar (A, C, G, I, L, M, F, P, W, V) are defined. Therefore 4 different labels are used to represent polar to polar, polar to nonpolar, nonpolar to polar and nonpolar to nonpolar change caused by mutation.
- Hydrogen bond: Label encoding method uses 16 different labels representing interchange between four classes: (i) donor (R, K, W) (ii) acceptor (D, E) (iii) donor and acceptor (N, Q, H, S, T, Y) and (iv) none (A, C, G, I, L, M, F, P, V) over mutation.
- Label hydrophobicity: Nine classes are labeled corresponding to interchange between three classes: (i) hydrophobic (A, C, I, L, M, F, W, V) (ii) Neutral (G, H, P, S, T, Y) and (iii) hydrophilic (R, N, D, Q, E, K) due to mutation.
4.3.3. Sequence-Based Features
4.3.4. Distance-Based Features
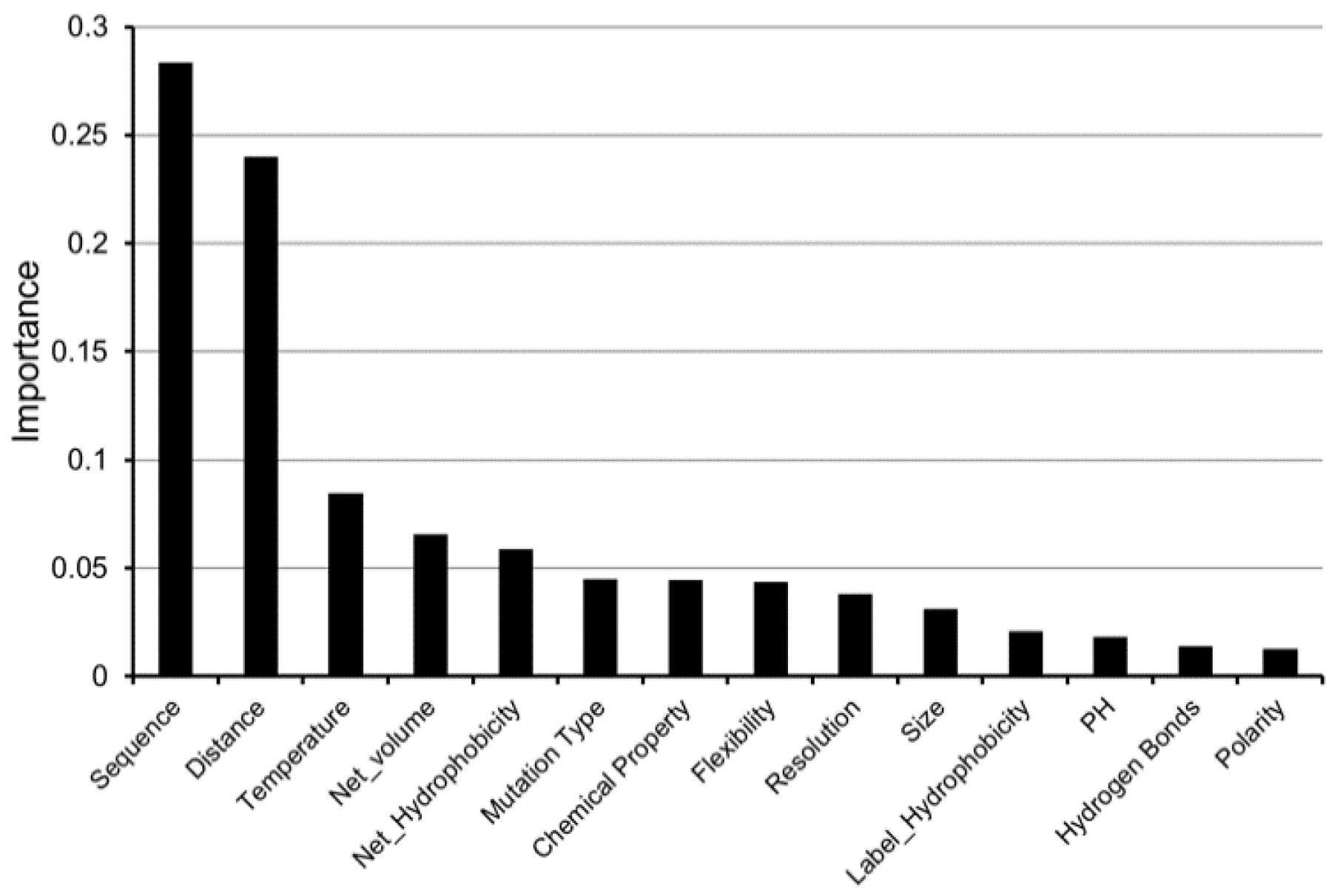
4.4. Feature Importance Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Braun, P.; Gingras, A.-C. History of protein–protein interactions: From egg-white to complex networks. Proteomics 2012, 12, 1478–1498. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA; Abingdon, UK, 2014. [Google Scholar]
- Ganeshpurkar, A.; Swetha, R.; Kumar, D.; Gangaram, G.P.; Singh, R.; Gutti, G.; Jana, S.; Kumar, D.; Kumar, A.; Singh, S.K. Protein-Protein Interactions and Aggregation Inhibitors in Alzheimer’s Disease. Curr. Top. Med. Chem. 2019, 19, 501–533. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.K.; Kurakula, K.; Koenis, D.S.; de Vries, C.J. Protein-protein interactions of the LIM-only protein FHL2 and functional implication of the interactions relevant in cardiovascular disease. Biochim. Biophys. Acta 2016, 1863, 219–228. [Google Scholar] [CrossRef]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefl, S.; Nishi, H.; Petukh, M.; Panchenko, A.R.; Alexov, E. Molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013, 425, 3919–3936. [Google Scholar] [CrossRef] [Green Version]
- Spevacek, A.R.; Evans, E.G.; Miller, J.L.; Meyer, H.C.; Pelton, J.G.; Millhauser, G.L. Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure 2013, 21, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Nishi, H.; Tyagi, M.; Teng, S.; Shoemaker, B.A.; Hashimoto, K.; Alexov, E.; Wuchty, S.; Panchenko, A.R. Cancer missense mutations alter binding properties of proteins and their interaction networks. PLoS ONE 2013, 8, e66273. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kucukkal, T.G.; Li, J.; Alexov, E.; Cao, W. Binding Analysis of Methyl-CpG Binding Domain of MeCP2 and Rett Syndrome Mutations. ACS Chem. Biol. 2016, 11, 2706–2715. [Google Scholar] [CrossRef]
- Kucukkal, T.G.; Yang, Y.; Uvarov, O.; Cao, W.; Alexov, E. Impact of Rett Syndrome Mutations on MeCP2 MBD Stability. Biochemistry 2015, 54, 6357–6368. [Google Scholar] [CrossRef]
- Yu, H.; Ye, L.; Wang, J.; Jin, L.; Lv, Y.; Yu, M. Protein-protein interaction networks and modules analysis for colorectal cancer and serrated adenocarcinoma. J. Cancer Res. 2015, 11, 846–851. [Google Scholar]
- Peng, Y.; Alexov, E. Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins 2016, 84, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On human disease-causing amino acid variants: Statistical study of sequence and structural patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Martiny, V.; Lagorce, D.; Ikeguchi, Y.; Alexov, E.; Miteva, M.A. Rational design of small-molecule stabilizers of spermine synthase dimer by virtual screening and free energy-based approach. PLoS ONE 2014, 9, e110884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Witham, S.; Petukh, M.; Moroy, G.; Miteva, M.; Ikeguchi, Y.; Alexov, E. A rational free energy-based approach to understanding and targeting disease-causing missense mutations. J. Am. Med. Inform. Assoc. 2013, 20, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, P.W.; Cole, K.; Gordon, N.C.; Kearns, A.M.; Llewelyn, M.J.; Peto, T.E.A.; Crook, D.W.; Walker, A.S. Robust Prediction of Resistance to Trimethoprim in Staphylococcus aureus. Cell Chem. Biol. 2018, 25, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Hauser, K.; Negron, C.; Albanese, S.K.; Ray, S.; Steinbrecher, T.; Abel, R.; Chodera, J.D.; Wang, L. Predicting resistance of clinical Abl mutations to targeted kinase inhibitors using alchemical free-energy calculations. Commun. Biol. 2018, 1, 70. [Google Scholar] [CrossRef] [Green Version]
- Griss, R.; Schena, A.; Reymond, L.; Patiny, L.; Werner, D.; Tinberg, C.E.; Baker, D.; Johnsson, K. Bioluminescent sensor proteins for point-of-care therapeutic drug monitoring. Nat. Chem. Biol. 2014, 10, 598–603. [Google Scholar] [CrossRef]
- Zhou, L.; Bosscher, M.; Zhang, C.; Özçubukçu, S.; Zhang, L.; Zhang, W.; Li, C.J.; Liu, J.; Jensen, M.P.; Lai, L.; et al. A protein engineered to bind uranyl selectively and with femtomolar affinity. Nat. Chem. 2014, 6, 236–241. [Google Scholar] [CrossRef]
- Yugandhar, K.; Gupta, S.; Yu, H. Inferring Protein-Protein Interaction Networks From Mass Spectrometry-Based Proteomic Approaches: A Mini-Review. Comput. Struct. Biotechnol. J. 2019, 17, 805–811. [Google Scholar] [CrossRef]
- Kao, F.S.; Ger, W.; Pan, Y.R.; Yu, H.C.; Hsu, R.Q.; Chen, H.M. Chip-based protein-protein interaction studied by atomic force microscopy. Biotechnol. Bioeng. 2012, 109, 2460–2467. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, K.S.; Bogan, A.A. ASEdb: A database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics 2001, 17, 284–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.D.S.; Gromiha, M.M. PINT: Protein–protein Interactions Thermodynamic Database. Nucleic Acids Res. 2006, 34, D195–D198. [Google Scholar] [CrossRef]
- Sirin, S.; Apgar, J.R.; Bennett, E.M.; Keating, A.E. AB-Bind: Antibody binding mutational database for computational affinity predictions. Protein Sci. 2016, 25, 393–409. [Google Scholar] [CrossRef] [Green Version]
- Jemimah, S.; Yugandhar, K.; Michael Gromiha, M. PROXiMATE: A database of mutant protein–protein complex thermodynamics and kinetics. Bioinformatics 2017, 33, 2787–2788. [Google Scholar] [CrossRef]
- Geng, C.; Vangone, A.; Bonvin, A.M.J.J. Exploring the interplay between experimental methods and the performance of predictors of binding affinity change upon mutations in protein complexes. Protein Eng. Des. Sel. 2016, 29, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Moal, I.H.; Fernández-Recio, J. SKEMPI: A Structural Kinetic and Energetic database of Mutant Protein Interactions and its use in empirical models. Bioinformatics 2012, 28, 2600–2607. [Google Scholar] [CrossRef] [Green Version]
- Jankauskaite, J.; Jiménez-García, B.; Dapkunas, J.; Fernández-Recio, J.; Moal, I.H. SKEMPI 2.0: An updated benchmark of changes in protein-protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 2019, 35, 462–469. [Google Scholar] [CrossRef]
- Vihinen, M. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genom. 2012, 13, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2013, 30, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brender, J.R.; Zhang, Y. Predicting the Effect of Mutations on Protein-Protein Binding Interactions through Structure-Based Interface Profiles. PLoS Comput. Biol. 2015, 11, e1004494. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein–protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petukh, M.; Li, M.; Alexov, E. Predicting Binding Free Energy Change Caused by Point Mutations with Knowledge-Modified MM/PBSA Method. PLoS Comput. Biol. 2015, 11, e1004276. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Simonetti, F.L.; Goncearenco, A.; Panchenko, A.R. MutaBind estimates and interprets the effects of sequence variants on protein–protein interactions. Nucleic Acids Res. 2016, 44, W494–W501. [Google Scholar] [CrossRef] [Green Version]
- Moal, I.H.; Fernandez-Recio, J. Intermolecular Contact Potentials for Protein–Protein Interactions Extracted from Binding Free Energy Changes upon Mutation. J. Chem. Theory Comput. 2013, 9, 3715–3727. [Google Scholar] [CrossRef]
- Niroula, A.; Vihinen, M. Variation Interpretation Predictors: Principles, Types, Performance, and Choice. Hum. Mutat. 2016, 37, 579–597. [Google Scholar] [CrossRef]
- Vihinen, M. Proper reporting of predictor performance. Nat. Methods 2014, 11, 781. [Google Scholar] [CrossRef] [Green Version]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortemme, T.; Baker, D. A simple physical model for binding energy hot spots in protein–protein complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 14116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedix, A.; Becker, C.M.; de Groot, B.L.; Caflisch, A.; Böckmann, R.A. Predicting free energy changes using structural ensembles. Nat. Methods 2009, 6, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Petukh, M.; Alexov, E.; Panchenko, A.R. Predicting the Impact of Missense Mutations on Protein-Protein Binding Affinity. J. Chem. Theory Comput. 2014, 10, 1770–1780. [Google Scholar] [CrossRef]
- Petukh, M.; Dai, L.; Alexov, E. SAAMBE: Webserver to Predict the Charge of Binding Free Energy Caused by Amino Acids Mutations. Int J. Mol. Sci 2016, 17, 547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, P.; Zhang, C.; Zheng, W.; Zhang, Y. BindProfX: Assessing Mutation-Induced Binding Affinity Change by Protein Interface Profiles with Pseudo-Counts. J. Mol. Biol. 2017, 429, 426–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, C.; Vangone, A.; Folkers, G.E.; Xue, L.C.; Bonvin, A.M.J.J. iSEE: Interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations. Proteins Struct. Funct. Bioinform. 2019, 87, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.H.M.; Myung, Y.; Pires, D.E.V.; Ascher, D.B. mCSM-PPI2: Predicting the effects of mutations on protein–protein interactions. Nucleic Acids Res. 2019, 47, W338–W344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Chen, Y.; Lu, H.; Zhao, F.; Alvarez, R.V.; Goncearenco, A.; Panchenko, A.R.; Li, M. MutaBind2: Predicting the impact of single and multiple point mutations on protein–protein interactions. iScience 2020, 100939, in press. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Cang, Z.; Wei, G.-W. A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation. Nat. Mach. Intell. 2020, 2, 116–123. [Google Scholar] [CrossRef]
- Wei, X.; Das, J.; Fragoza, R.; Liang, J.; Bastos de Oliveira, F.M.; Lee, H.R.; Wang, X.; Mort, M.; Stenson, P.D.; Cooper, D.N.; et al. A Massively Parallel Pipeline to Clone DNA Variants and Examine Molecular Phenotypes of Human Disease Mutations. PLoS Genetics 2014, 10, e1004819. [Google Scholar] [CrossRef]
- Fragoza, R.; Das, J.; Wierbowski, S.D.; Liang, J.; Tran, T.N.; Liang, S.; Beltran, J.F.; Rivera-Erick, C.A.; Ye, K.; Wang, T.-Y.; et al. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun. 2019, 10, 4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, D.; Xie, T.-X.; Ow, T.J.; Zhao, M.; Pickering, C.R.; Zhou, G.; Sandulache, V.C.; Wheeler, D.A.; Gibbs, R.A.; Caulin, C.; et al. Disruptive TP53 mutation is associated with aggressive disease characteristics in an orthotopic murine model of oral tongue cancer. Clin. Cancer Res. 2011, 17, 6658–6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Chen, P.; Wang, B.; Zhang, J.; Li, J. dbMPIKT: A database of kinetic and thermodynamic mutant protein interactions. BMC Bioinform. 2018, 19, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petukh, M.; Stefl, S.; Alexov, E. The role of protonation states in ligand-receptor recognition and binding. Curr. Pharm. Des. 2013, 19, 4182–4190. [Google Scholar] [CrossRef] [Green Version]
- Alexov, E. Calculating proton uptake/release and binding free energy taking into account ionization and conformation changes induced by protein–inhibitor association: Application to plasmepsin, cathepsin D and endothiapepsin–pepstatin complexes. Proteins Struct. Funct. Bioinform. 2004, 56, 572–584. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Alexov, E. Protonation and pK changes in protein-ligand binding. Q. Rev. Biophys. 2013, 46, 181–209. [Google Scholar] [CrossRef] [Green Version]
- Moon, C.P.; Fleming, K.G. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc. Natl. Acad. Sci. USA 2011, 108, 10174–10177. [Google Scholar] [CrossRef] [Green Version]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stabilizing and Destabilizing | Highly Stabilizing and Destabilizing | |
|---|---|---|
| AUC | 0.75 | 0.99 |
| Sensitivity | 0.82 | 0.95 |
| Specificity | 0.53 | 1 |
| Precision | 0.86 | 1 |
| Accuracy | 0.76 | 0.96 |
| MCC | 0.34 | 0.82 |
| Homo-Dimer | Hetero-Dimer | |
|---|---|---|
| AUC | 1 | 0.96 |
| Sensitivity | 0.99 | 1 |
| Specificity | 0.99 | 0.95 |
| Precision | 1 | 0.8 |
| Accuracy | 1 | 0.96 |
| Method | Time of Calculation |
|---|---|
| mCSM-PPI2 | 42 seconds |
| SAAMBE-3D | 0.21 seconds |
| MutaBind2 | 10 minutes |
| BindProfX | 50 minutes |
| BeAtMuSiC | 2 seconds |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pahari, S.; Li, G.; Murthy, A.K.; Liang, S.; Fragoza, R.; Yu, H.; Alexov, E. SAAMBE-3D: Predicting Effect of Mutations on Protein–Protein Interactions. Int. J. Mol. Sci. 2020, 21, 2563. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072563
Pahari S, Li G, Murthy AK, Liang S, Fragoza R, Yu H, Alexov E. SAAMBE-3D: Predicting Effect of Mutations on Protein–Protein Interactions. International Journal of Molecular Sciences. 2020; 21(7):2563. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072563
Chicago/Turabian StylePahari, Swagata, Gen Li, Adithya Krishna Murthy, Siqi Liang, Robert Fragoza, Haiyuan Yu, and Emil Alexov. 2020. "SAAMBE-3D: Predicting Effect of Mutations on Protein–Protein Interactions" International Journal of Molecular Sciences 21, no. 7: 2563. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072563