Interpretation of the Epigenetic Signature of Facioscapulohumeral Muscular Dystrophy in Light of Genotype-Phenotype Studies

 , , ,
, , ,  , , , ,
, , , ,  , , add
Show full author list
, , add
Show full author list
Abstract
:1. Introduction
2. Results
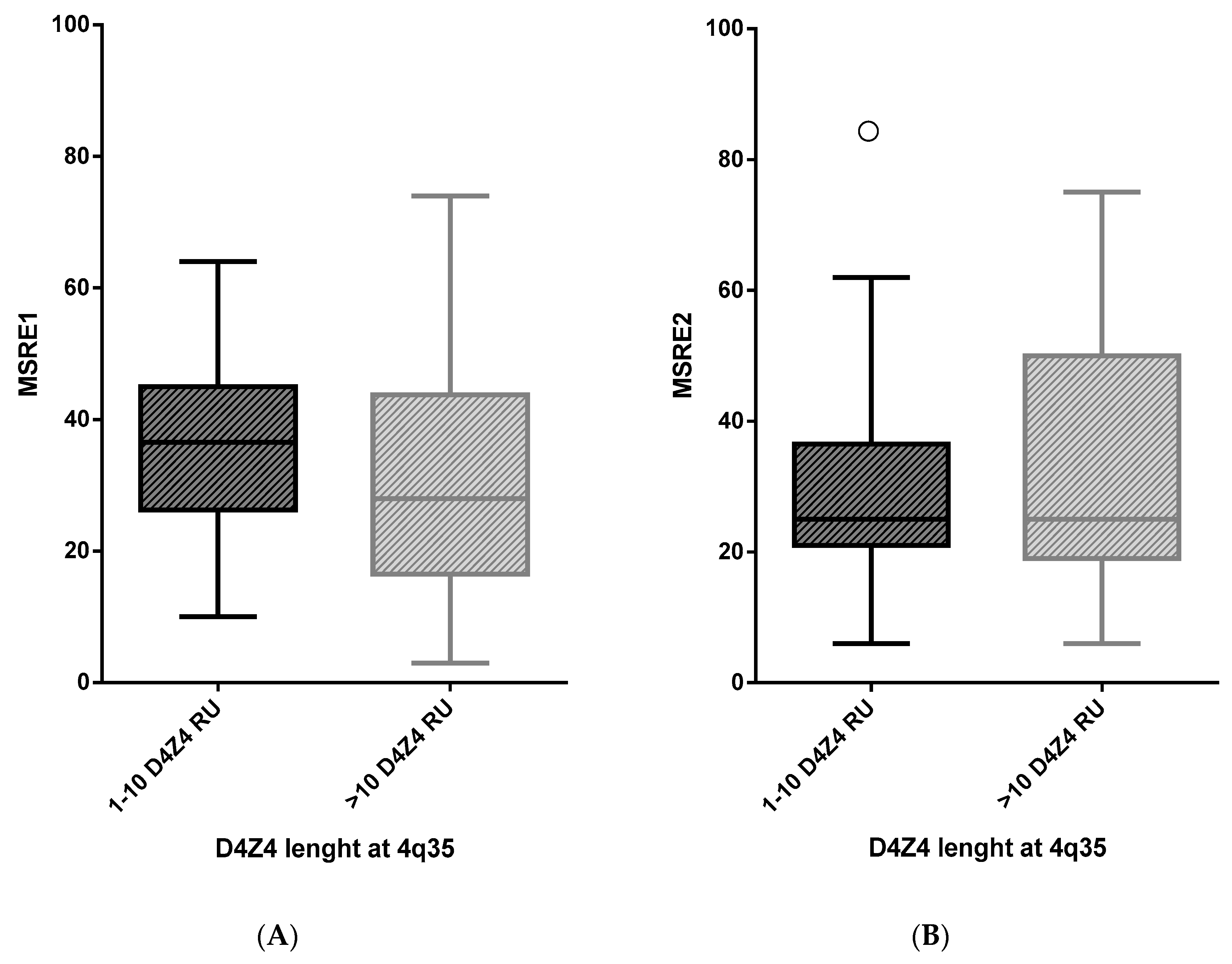
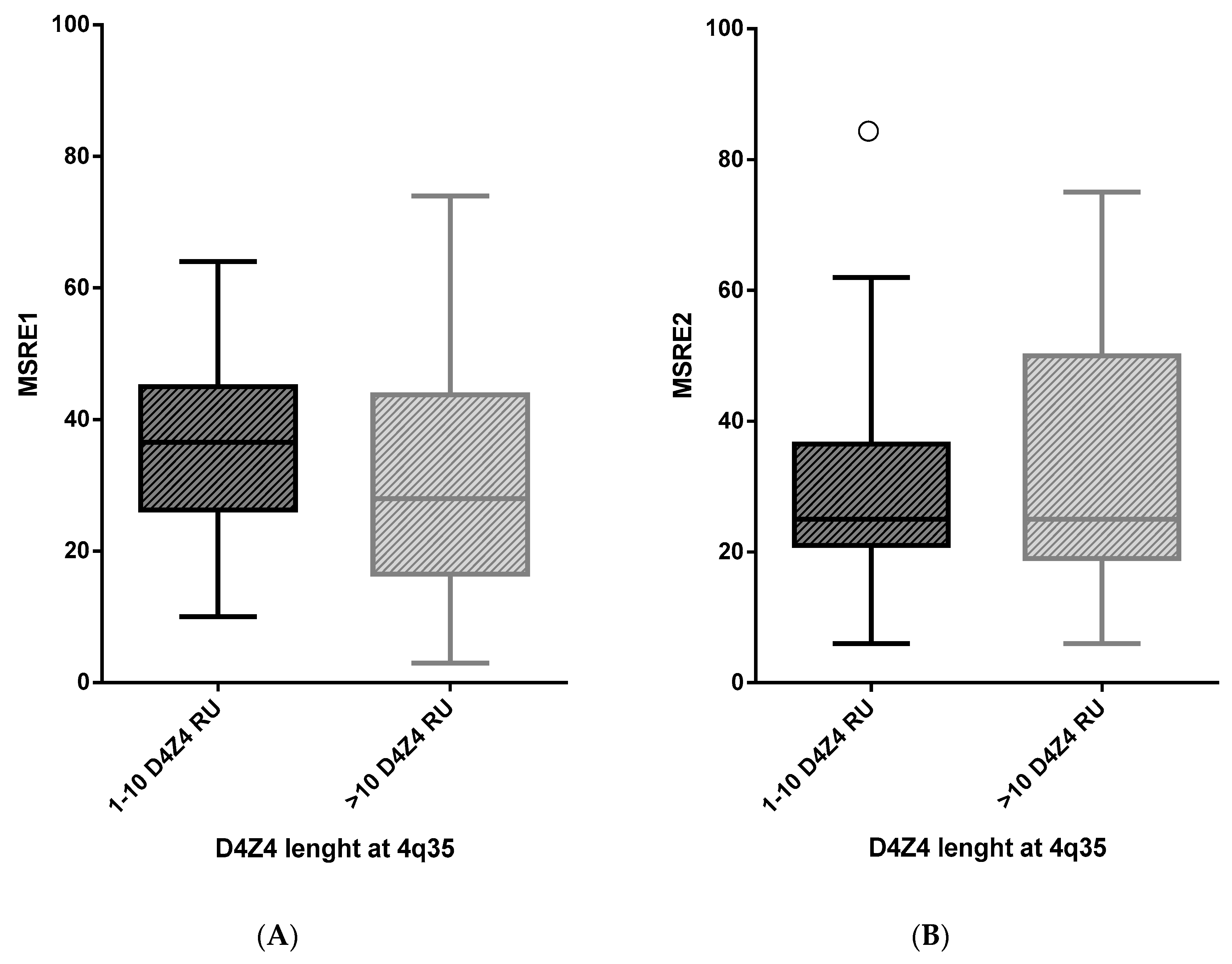
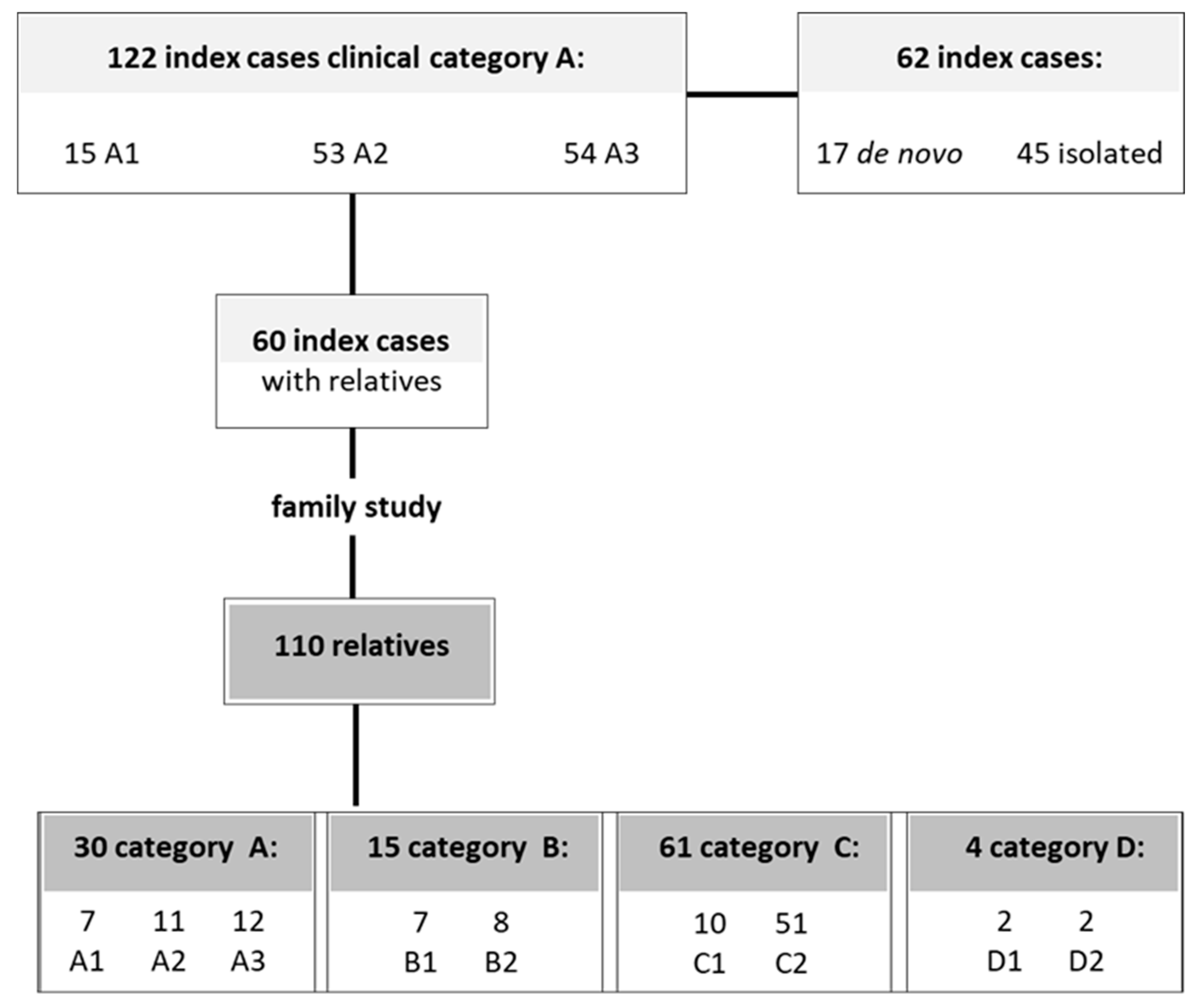
2.1. A Highly Variable Range of D4Z4 Methylation Level in FSHD ndex Cases
2.2. Analysis of D4Z4 Methylation in FSHD1 and FSHD2
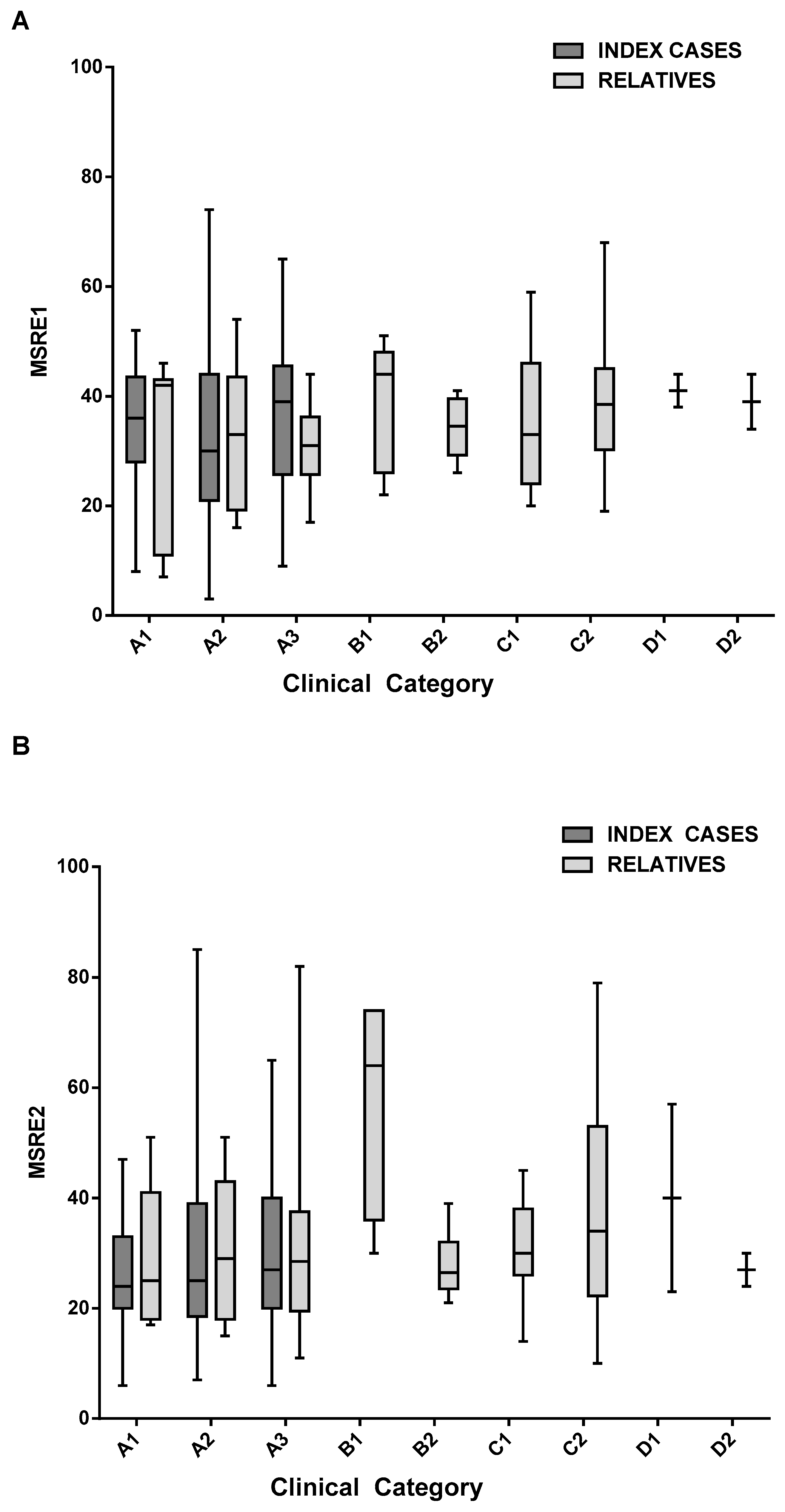
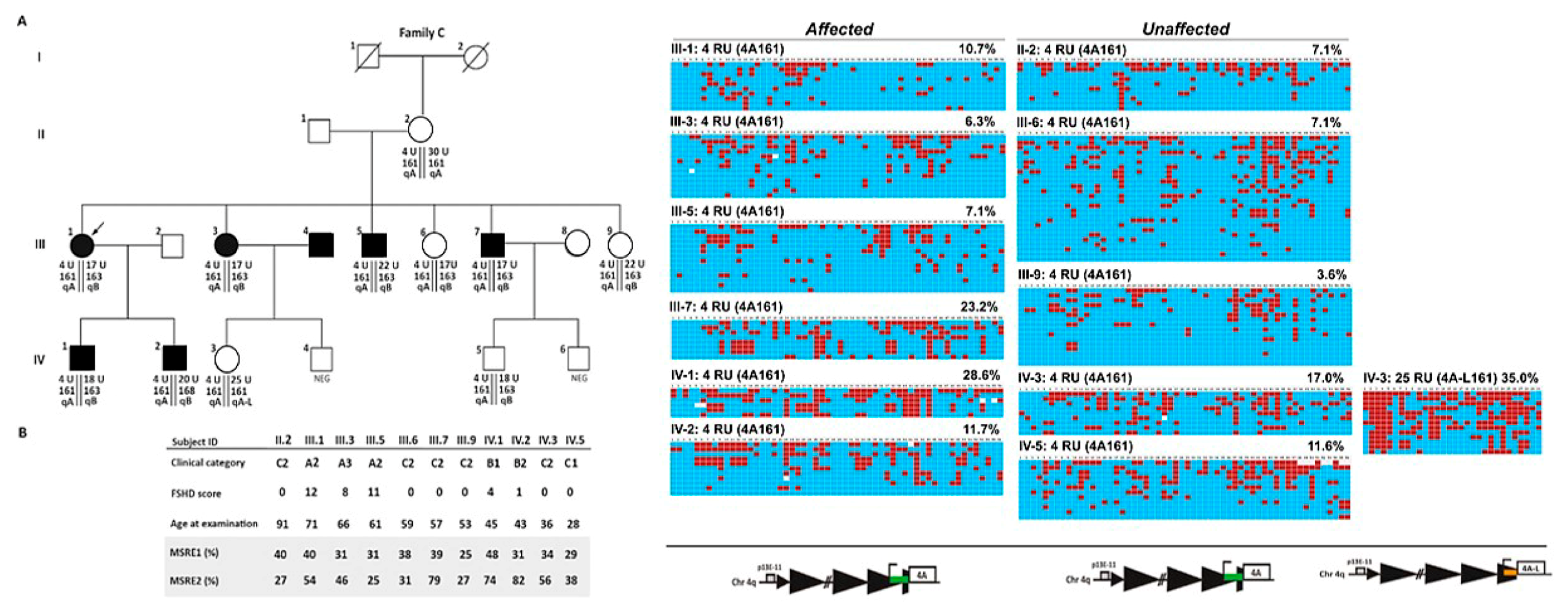
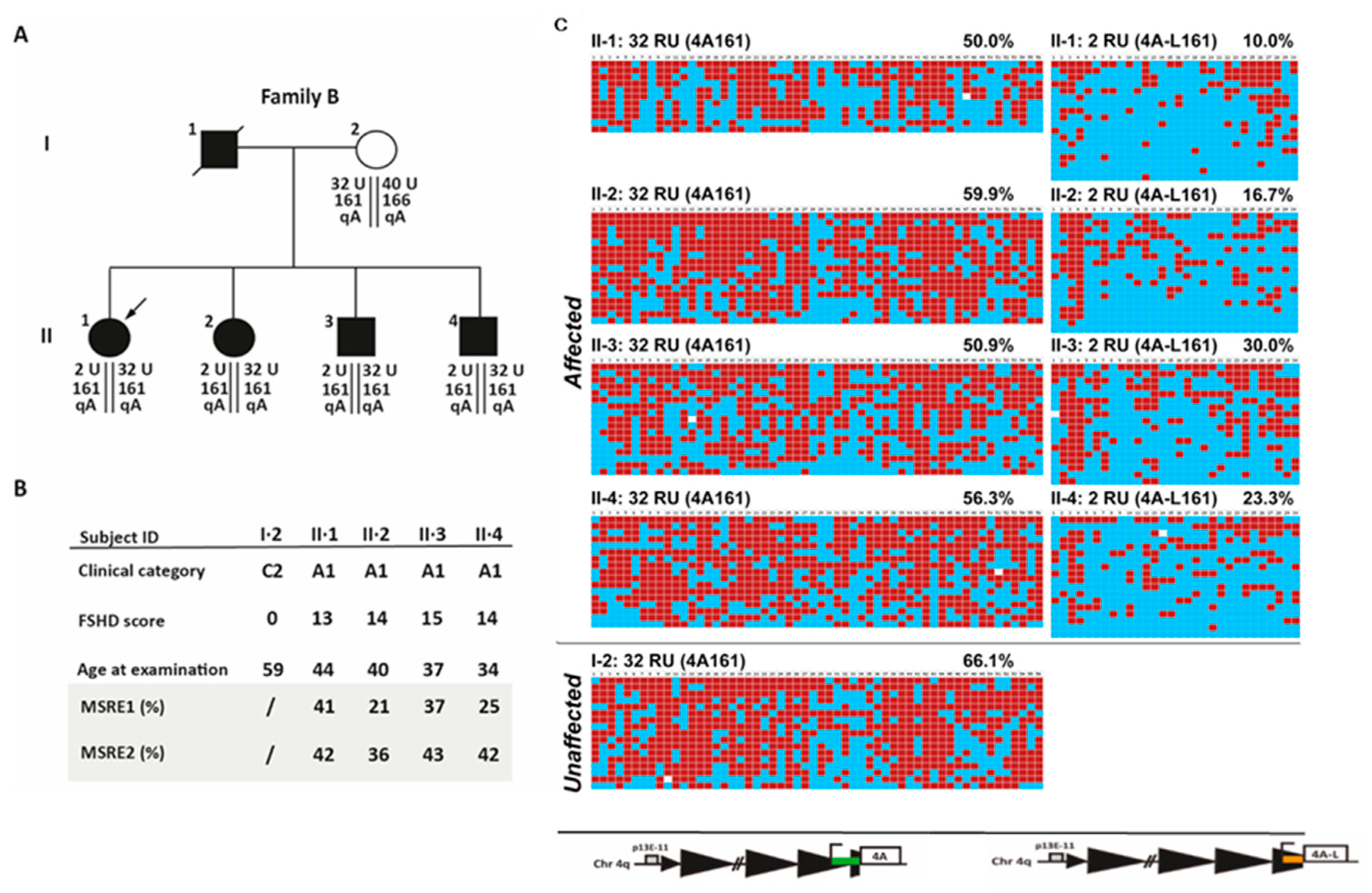
2.3. Analysis of D4Z4 DNA Methylation in FSHD Families
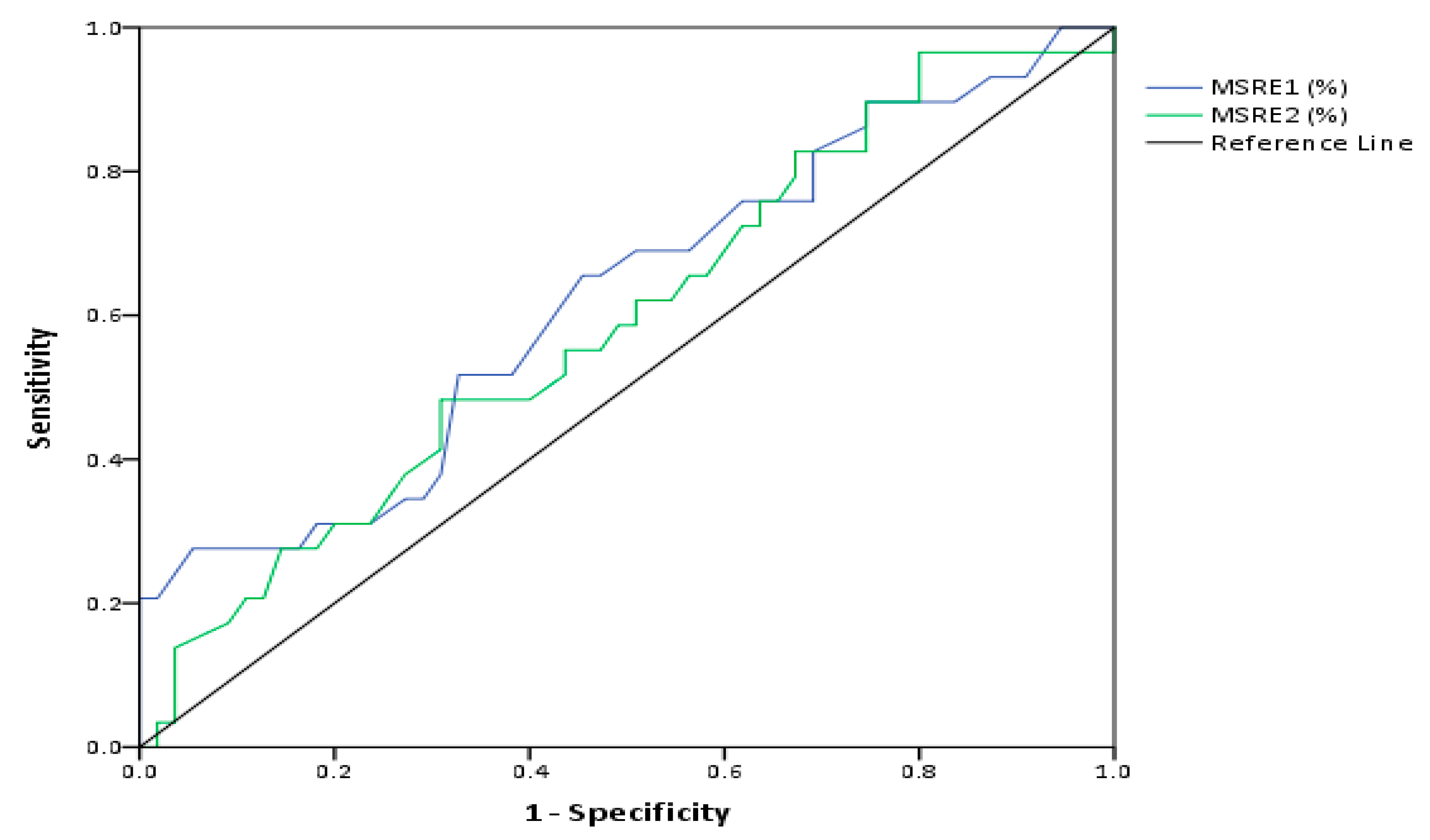
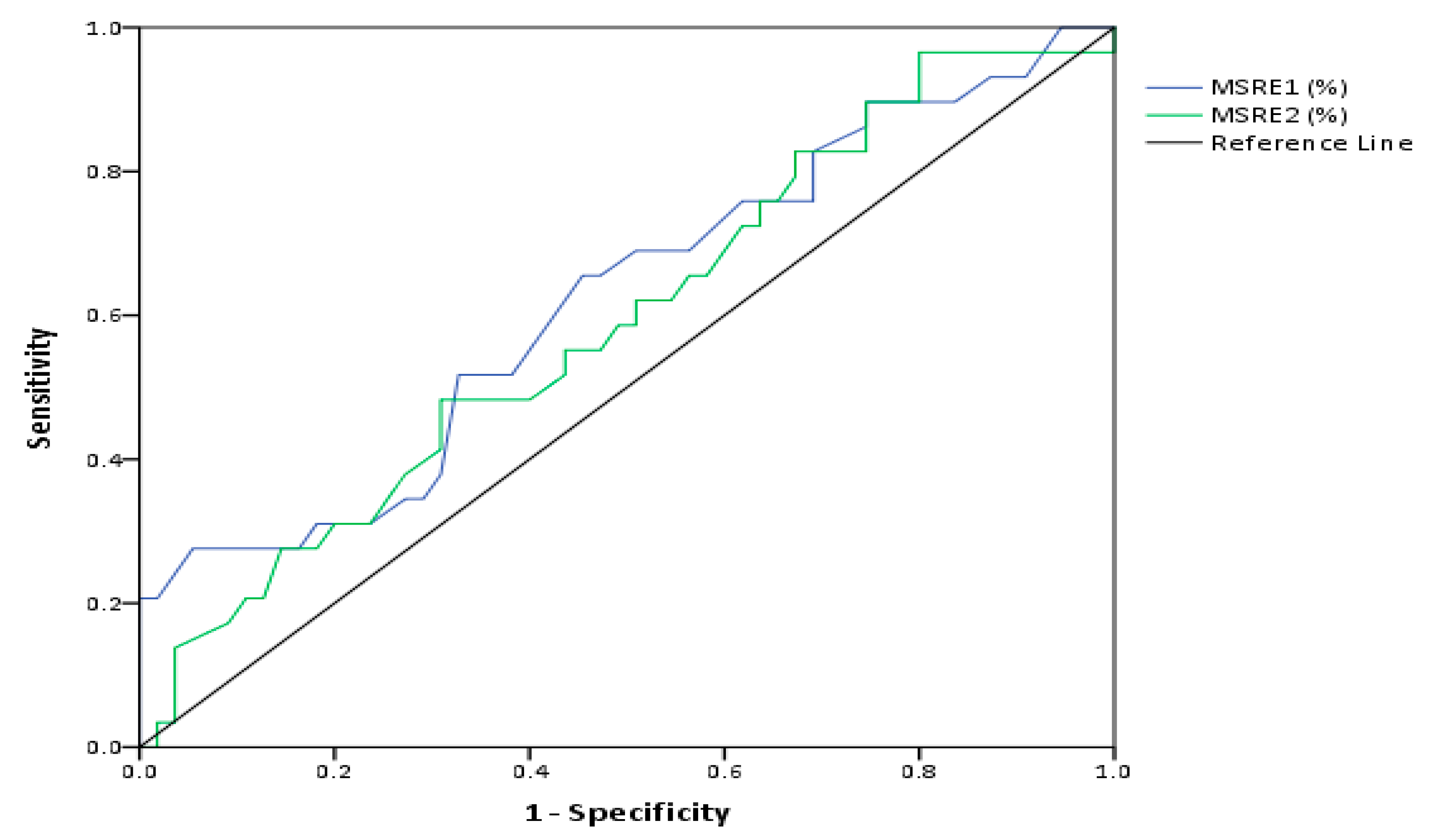
2.4. Evaluation of the Predictive Value of D4Z4 DNA Methylation in FSHD
2.5. Analysis of D4Z4 Methylation Status and Reduced Penetrance in FSHD Families
2.6. D4Z4 DNA Methylation Analysis in A Fully Penetrant FSHD1 Family
3. Discussion
4. Materials and Methods
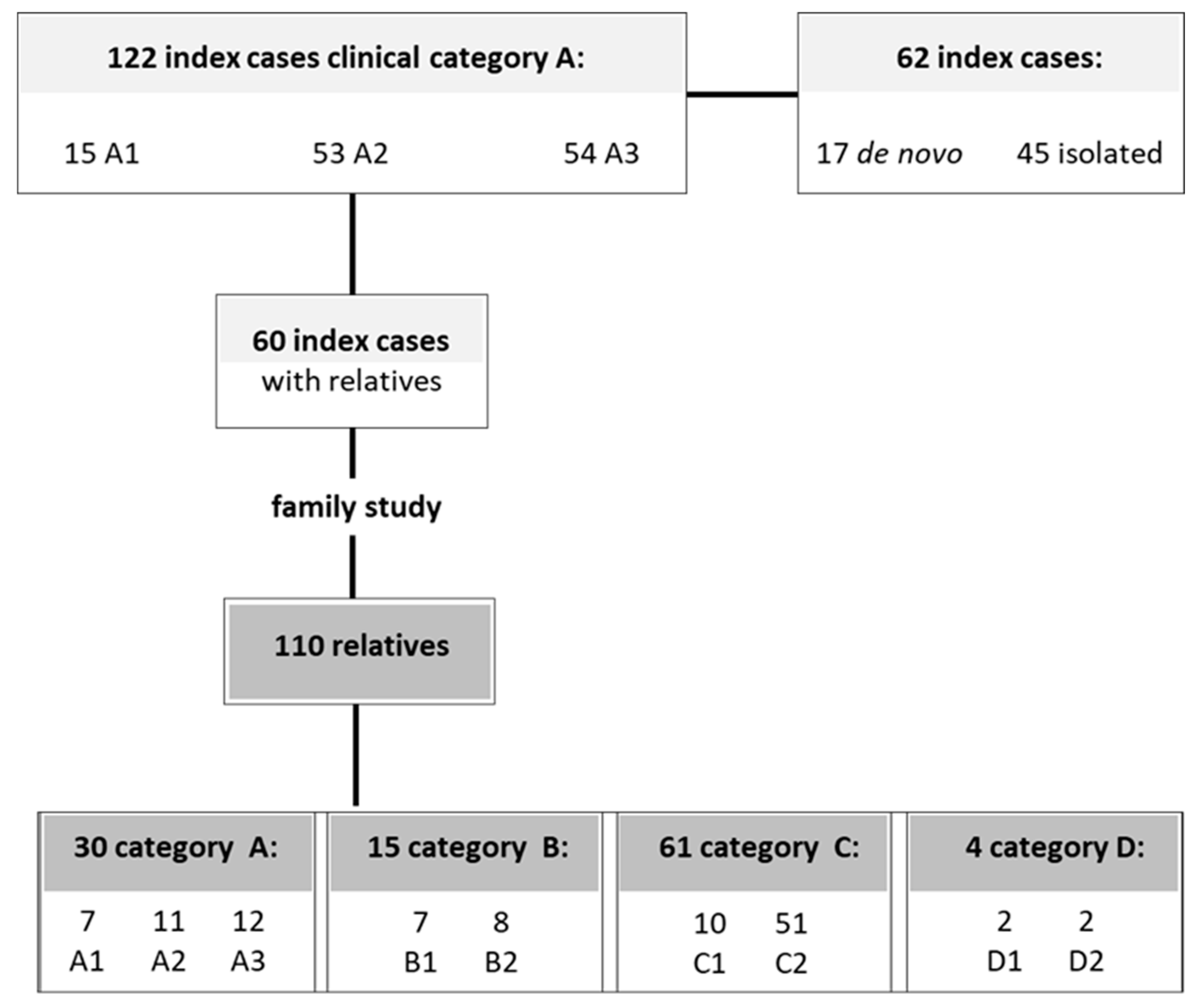
4.1. Participants
4.2. Clinical Investigation
4.3. Molecular Analysis for 4q35 and 10q26
4.4. Methylation-Sensitive Restriction Enzyme (MSRE) Analysis
4.5. Sodium Bisulfite Sequencing (BSS)-DNA Methylation Analysis
4.6. Statistical Analysis
4.7. Ethical Statement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Padberg, G.W.A.M. Facioscapulohumeral Disease; Leiden University: Leiden, The Netherlands, 1982. [Google Scholar]
- Mostacciuolo, M.L.; Pastorello, E.; Vazza, G.; Miorin, M.; Angelini, C.; Tomelleri, G.; Galluzzi, G.; Trevisan, C.P. Facioscapulohumeral muscular dystrophy: Epidemiological and molecular study in a north-east Italian population sample. Clin. Genet. 2009, 75, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Nishino, I.; Hayashi, Y.K. Very low penetrance in 85 Japanese families with facioscapulohumeral muscular dystrophy 1A. J. Med. Genet. 2004, 41, e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, M.; Matsuzaki, T.; Higuchi, I.; Fukunaga, H.; Inui, T.; Nagamitsu, S.; Yamada, H.; Arimura, K.; Osame, M. Facioscapulohumeral Muscular Dystrophy: Clinical Diversity and Genetic Abnormalities in Japanese Patients. Intern. Med. 1997, 36, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, T.I.; Chen, J.C.J.; Rahimov, F.; Homma, S.; Arashiro, P.; Beermann, M.L.; King, O.D.; Miller, J.B.; Kunkel, L.M.; Emerson, C.P.; et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: Evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 2012, 21, 4419–4430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.-M.; Hofker, M.H.; Moerer, P.; Williamson, R.; et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992, 2, 26–30. [Google Scholar] [CrossRef]
- Deutekom, J.C.T.V.; Wljmenga, C.; Tlenhoven, E.A.E.V.; Gruter, A.-M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.-J.B.; Hofker, M.H.; Fronts, R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Deidda, G.; Cacurri, S.; Piazzo, N.; Felicetti, L. Direct detection of 4q35 rearrangements implicated in facioscapulohumeral muscular dystrophy (FSHD). J. Med. Genet. 1996, 33, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Lunt, P. 44th ENMC International Workshop: Facioscapulohumeral Muscular Dystrophy: Molecular Studies: 19–21 July 1996, Naarden, The Netherlands. Neuromuscul. Disord. 1998, 8, 126–130. [Google Scholar] [CrossRef]
- Scionti, I.; Greco, F.; Ricci, G.; Govi, M.; Arashiro, P.; Vercelli, L.; Berardinelli, A.; Angelini, C.; Antonini, G.; Cao, M.; et al. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2012, 90, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, M.; Maynard, J.; Rogers, M.T.; Lunt, P.W.; Jardine, P.; Ravine, D.; Harper, P.S. Improved molecular diagnosis of facioscapulohumeral muscular dystrophy (FSHD): Validation of the differential double digestion for FSHD. J. Med. Genet. 1997, 34, 476–479. [Google Scholar] [CrossRef] [Green Version]
- de Greef, J.C.; Lemmers, R.J.L.F.; Camano, P.; Day, J.W.; Sacconi, S.; Dunand, M.; van Engelen, B.G.M.; Kiuru-Enari, S.; Padberg, G.W.; Rosa, A.L.; et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 2010, 75, 1548–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butz, M.; Koch, M.C.; Muller-Felber, W.; Lemmers, R.J.L.F.; van der Maarel, S.M.; Schreiber, H. Facioscapulohumeral muscular dystrophy. J. Neurol. 2003, 250, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, M.; Lemmers, R.J.; van der Kooi, E.L.; van der Wielen, M.J.; van Overveld, P.G.; Dauwerse, H.; Bakker, E.; Frants, R.R.; Padberg, G.W.; van der Maarel, S.M. Possible phenotypic dosage effect in patients compound heterozygous for FSHD-sized 4q35 alleles. Neurology 2003, 61, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.; Rost, S.; El Hajj, N.; Ferbert, A.; Deschauer, M.; Walter, M.C.; Schoser, B.; Tacik, P.; Kress, W.; Müller, C.R. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur. J. Hum. Genet. 2015, 23, 808–816. [Google Scholar] [CrossRef]
- Ricci, G.; Scionti, I.; Sera, F.; Govi, M.; D’Amico, R.; Frambolli, I.; Mele, F.; Filosto, M.; Vercelli, L.; Ruggiero, L.; et al. Large scale genotype–phenotype analyses indicate that novel prognostic tools are required for families with facioscapulohumeral muscular dystrophy. Brain 2013, 136, 3408–3417. [Google Scholar] [CrossRef] [Green Version]
- Ricci, G.; Zatz, M.; Tupler, R. Facioscapulohumeral Muscular Dystrophy: More Complex than it Appears. Curr. Mol. Med. 2014, 14, 1052–1068. [Google Scholar] [CrossRef]
- Salort-Campana, E.; Nguyen, K.; Bernard, R.; Jouve, E.; Solé, G.; Nadaj-Pakleza, A.; Niederhauser, J.; Charles, E.; Ollagnon, E.; Bouhour, F.; et al. Low penetrance in facioscapulohumeral muscular dystrophy type 1 with large pathological D4Z4 alleles: A cross-sectional multicenter study. Orphanet J. Rare Dis. 2015, 10, 2. [Google Scholar] [CrossRef]
- Ricci, G.; Ruggiero, L.; Vercelli, L.; Sera, F.; Nikolic, A.; Govi, M.; Mele, F.; Daolio, J.; Angelini, C.; Antonini, G.; et al. A novel clinical tool to classify facioscapulohumeral muscular dystrophy phenotypes. J. Neurol. 2016, 263, 1204–1214. [Google Scholar] [CrossRef]
- van Overveld, P.G.M.; Lemmers, R.J.F.L.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.-J.B.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003, 35, 315–317. [Google Scholar] [CrossRef]
- de Greef, J.C.; Lemmers, R.J.L.F.; van Engelen, B.G.M.; Sacconi, S.; Venance, S.L.; Frants, R.R.; Tawil, R.; van der Maarel, S.M. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum. Mutat. 2009, 30, 1449–1459. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.L.F.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.E.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balog, J.; Thijssen, P.E.; Shadle, S.; Straasheijm, K.R.; van der Vliet, P.J.; Krom, Y.D.; van den Boogaard, M.L.; de Jong, A.; F Lemmers, R.J.L.; Tawil, R.; et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics 2015, 10, 1133–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calandra, P.; Cascino, I.; Lemmers, R.J.L.F.L.F.; Galluzzi, G.; Teveroni, E.; Monforte, M.; Tasca, G.; Ricci, E.; Moretti, F.; Van Der Maarel, S.M.; et al. Allele-specific DNA hypomethylation characterises FSHD1 and FSHD2. J. Med. Genet. 2016, 53, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Hartweck, L.M.; Anderson, L.J.; Lemmers, R.J.; Dandapat, A.; Toso, E.A.; Dalton, J.C.; Tawil, R.; Day, J.W.; van der Maarel, S.M.; Kyba, M. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology 2013, 80, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, M.-C.; Roche, S.; Dion, C.; Tasmadjian, A.; Bouget, G.; Salort-Campana, E.; Vovan, C.; Chaix, C.; Broucqsault, N.; Morere, J.; et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 2014, 83, 733–742. [Google Scholar] [CrossRef]
- Jones, T.I.; Yan, C.; Sapp, P.C.; McKenna-Yasek, D.; Kang, P.B.; Quinn, C.; Salameh, J.S.; King, O.D.; Jones, P.L. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin. Epigenetics 2014, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.L.F.; Goeman, J.J.; van der Vliet, P.J.; van Nieuwenhuizen, M.P.; Balog, J.; Vos-Versteeg, M.; Camano, P.; Ramos Arroyo, M.A.; Jerico, I.; Rogers, M.T.; et al. A standardized clinical evaluation of patients affected by facioscapulohumeral muscular dystrophy: The FSHD clinical score. Muscle Nerve 2010, 42, 213–217. [Google Scholar]
- Jones, T.I.; King, O.D.; Himeda, C.L.; Homma, S.; Chen, J.C.J.J.; Beermann, M.L.; Yan, C.; Emerson, C.P.; Miller, J.B.; Wagner, K.R.; et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 2015, 24, 659–669. [Google Scholar]
- Jones, T.I.; Himeda, C.L.; Perez, D.P.; Jones, P.L. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin. Epigenetics 2015, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.I.; Himeda, C.L.; Perez, D.P.; Jones, P.L. Large family cohorts of lymphoblastoid cells provide a new cellular model for investigating facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2017, 27, 221–238. [Google Scholar] [CrossRef] [Green Version]
- Van Overveld, P.G.M.; Enthoven, L.; Ricci, E.; Rossi, M.; Felicetti, L.; Jeanpierre, M.; Winokur, S.T.; Frants, R.R.; Padberg, G.W.; Van Der Maarel, S.M. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar]
- de Greef, J.C.; Wohlgemuth, M.; Chan, O.A.; Hansson, K.B.; Smeets, D.; Frants, R.R.; Weemaes, C.M.; Padberg, G.W.; van der Maarel, S.M. Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann. Neurol. 2005, 58, 569–576. [Google Scholar]
- de Greef, J.C.; Frants, R.R.; van der Maarel, S.M. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology 2007, 69, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- de Greef, J.C.; Frants, R.R.; van der Maarel, S.M. Epigenetic mechanisms of facioscapulohumeral muscular dystrophy. Mutat. Res. 2008, 647, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FSHD Index Cases | D4Z4 (RU) | Number of Subjects | Mean D4Z4 Methylation Level (%) | SD | SE | Difference in Mean D4Z4 | p-Value |
|---|---|---|---|---|---|---|---|
| Methylation between Two Groups | |||||||
| Δ | |||||||
| MSRE1 | 1–10 | 88 | 36.3 | 11.9 | 1.3 | 4.7 | 0.108 |
| >10 | 34 | 31.6 | 19.5 | 3.3 | |||
| MSRE2 | 1–10 | 82 | 28.0 | 12.0 | 1.3 | −3.6 | 0.275 |
| >10 | 28 | 31.6 | 21.5 | 4.1 |
| MSRE1 | Clinical Category | Mean D4Z4 Methylation Level | 95% CI |
|---|---|---|---|
| index cases | A1 | 34.3 | [28, 40.6] |
| A2 | 33.2 | [33.2, 37.6] | |
| A3 | 37 | [33.4, 40.8] | |
| relatives | A1 | 33 | [16.2, 49.8] |
| A2 | 33.2 | [23.5, 42.9] | |
| A3 | 31.1 | [26.4, 35.9] | |
| B1 | 37.3 | [26.1, 48.5] | |
| B2 | 34.1 | [29.5, 38.7] | |
| C1 | 37.1 | [27.7, 46.5] | |
| C2 | 38.3 | [35.2, 41.4] | |
| D1 | 41 | [35.2, 46.8] | |
| D2 | 39 | [29.2, 48.8] |
| Test Result Variables | AUC | SE | Asymptotic 95% CI |
|---|---|---|---|
| MSRE1 | 0.622 | 0.1 | [0.5,0.8] |
| MSRE2 | 0.592 | 0.1 | [0.5,0.7] |
| Number of Subjects | Sex | Age at Examination | D4Z4 (RU) | ||||
|---|---|---|---|---|---|---|---|
| male | female | mean | SD | 1–10 | >10 | ||
| index cases | 122 | 73 (59.8%) | 49 (40.1%) | 48.5 | 18.5 | 88 | 34 |
| relatives | 110 | 49 (44.5%) | 61 (55.4%) | 45.3 | 15.5 | 107 | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolic, A.; Jones, T.I.; Govi, M.; Mele, F.; Maranda, L.; Sera, F.; Ricci, G.; Ruggiero, L.; Vercelli, L.; Portaro, S.; et al. Interpretation of the Epigenetic Signature of Facioscapulohumeral Muscular Dystrophy in Light of Genotype-Phenotype Studies. Int. J. Mol. Sci. 2020, 21, 2635. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072635
Nikolic A, Jones TI, Govi M, Mele F, Maranda L, Sera F, Ricci G, Ruggiero L, Vercelli L, Portaro S, et al. Interpretation of the Epigenetic Signature of Facioscapulohumeral Muscular Dystrophy in Light of Genotype-Phenotype Studies. International Journal of Molecular Sciences. 2020; 21(7):2635. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072635
Chicago/Turabian StyleNikolic, Ana, Takako I Jones, Monica Govi, Fabiano Mele, Louise Maranda, Francesco Sera, Giulia Ricci, Lucia Ruggiero, Liliana Vercelli, Simona Portaro, and et al. 2020. "Interpretation of the Epigenetic Signature of Facioscapulohumeral Muscular Dystrophy in Light of Genotype-Phenotype Studies" International Journal of Molecular Sciences 21, no. 7: 2635. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072635