
BRAF Modulates Stretch-Induced Intercellular Gap Formation through Localized Actin Reorganization

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
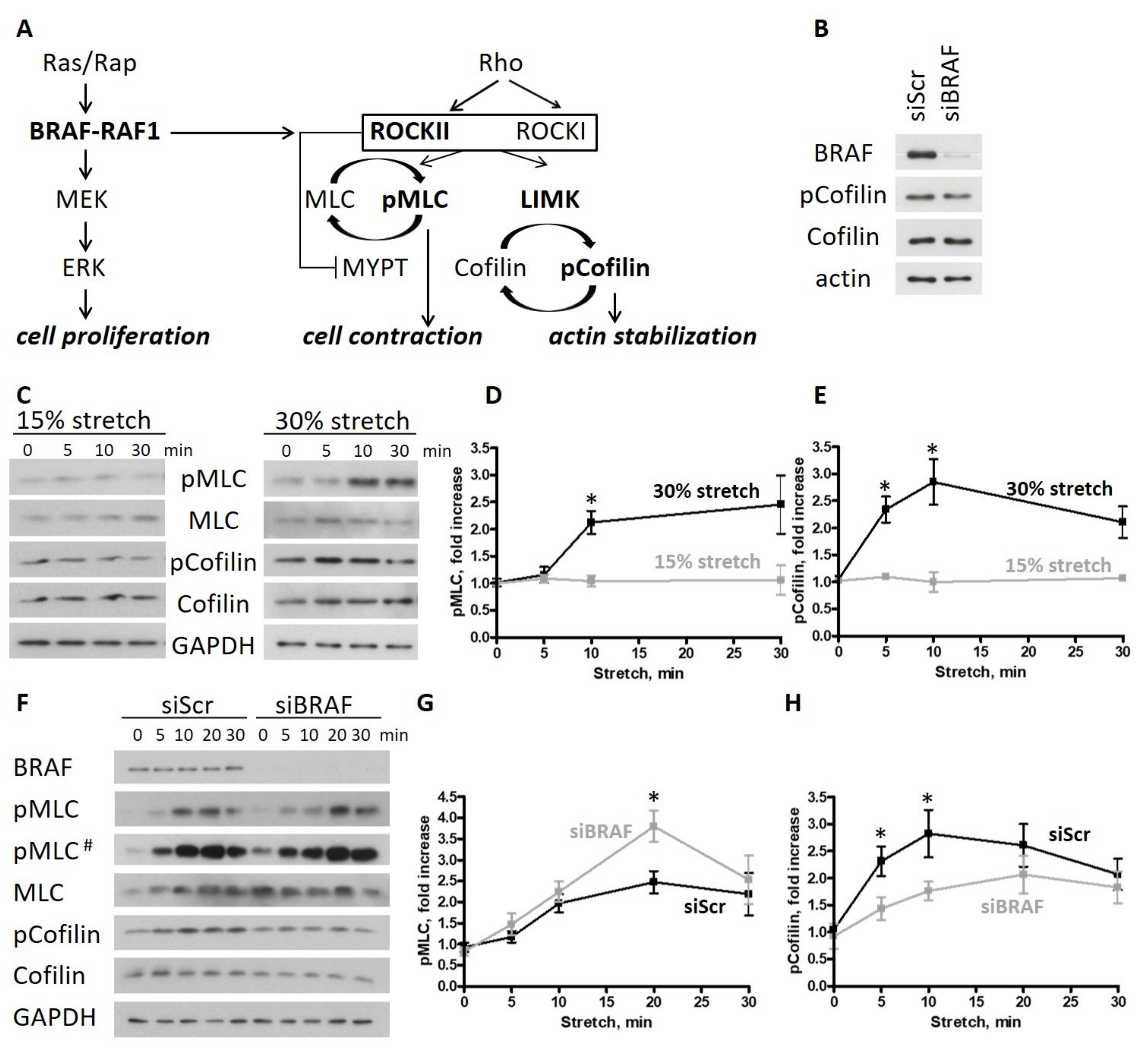
2.1. BRAF Knockdown Increases Cell Contractility and Actin Turnover upon Stretch in Endothelial Monolayers
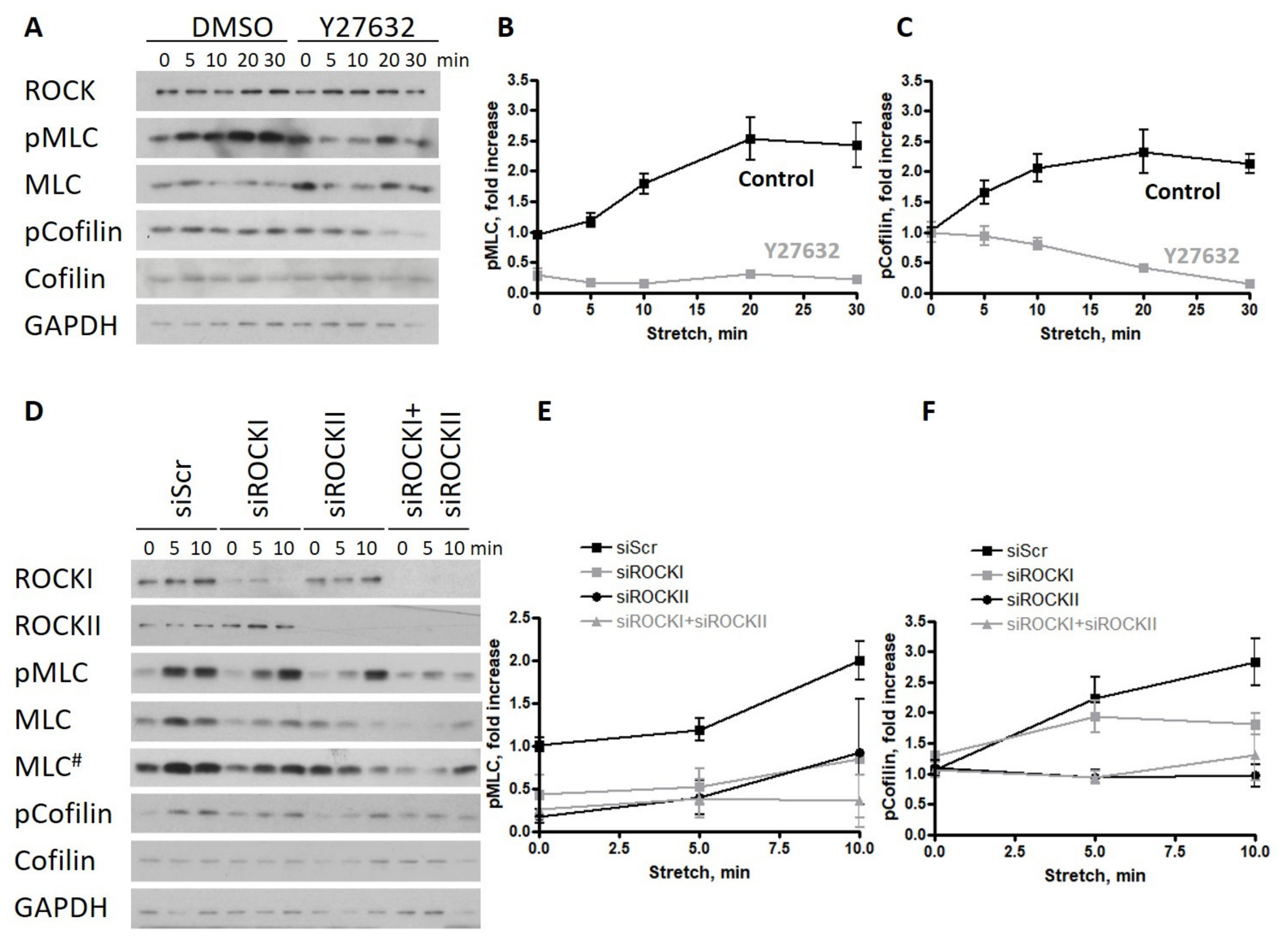
2.2. ROCKII Is Required for the Phosphorylation of Cofilin upon Stretch in Endothelial Monolayers
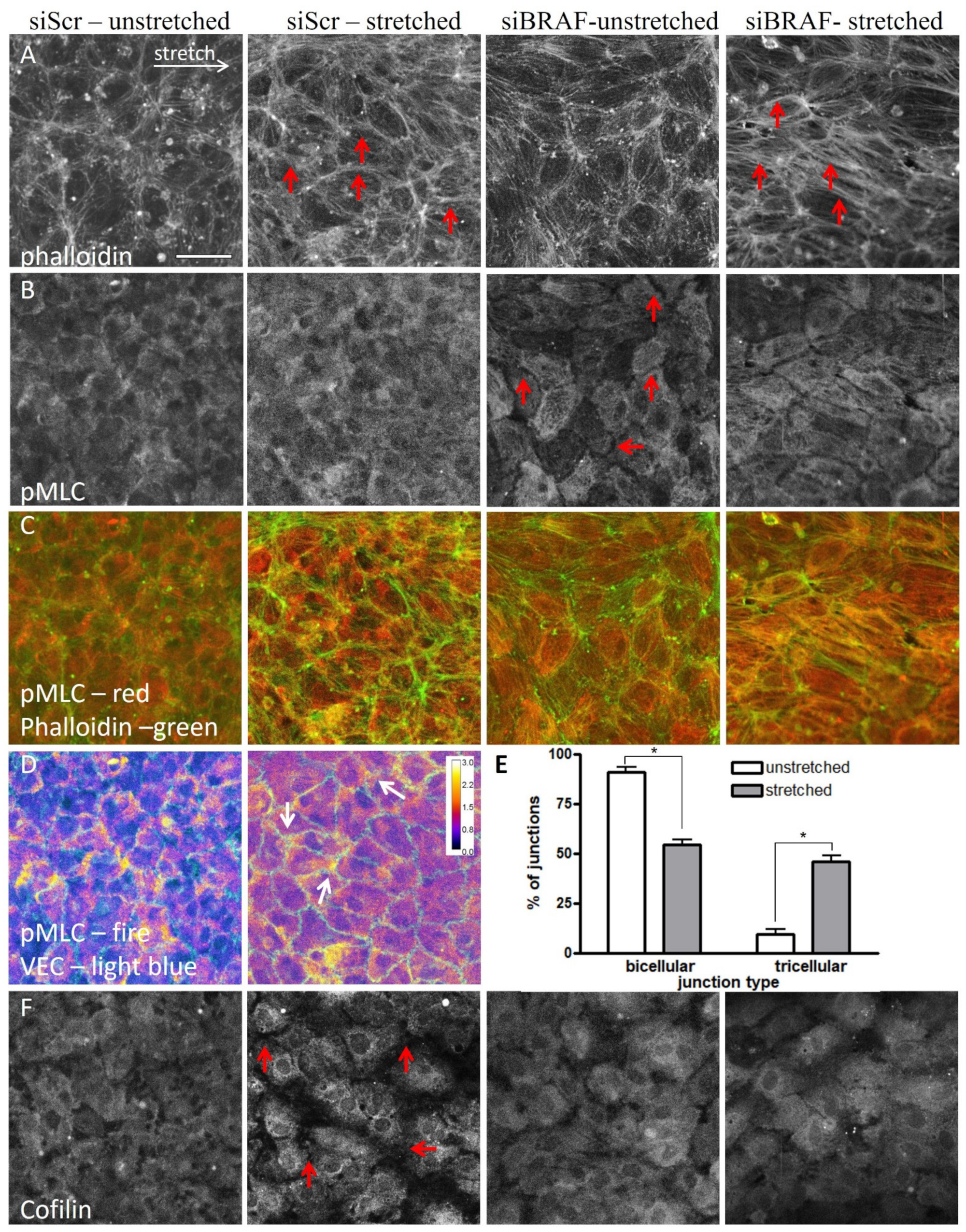
2.3. The Presence of BRAF Accelerates the Stabilization of the Newly Formed Actin Stress Fibers after Stretch in the Stretched (Horizontal) Junctions
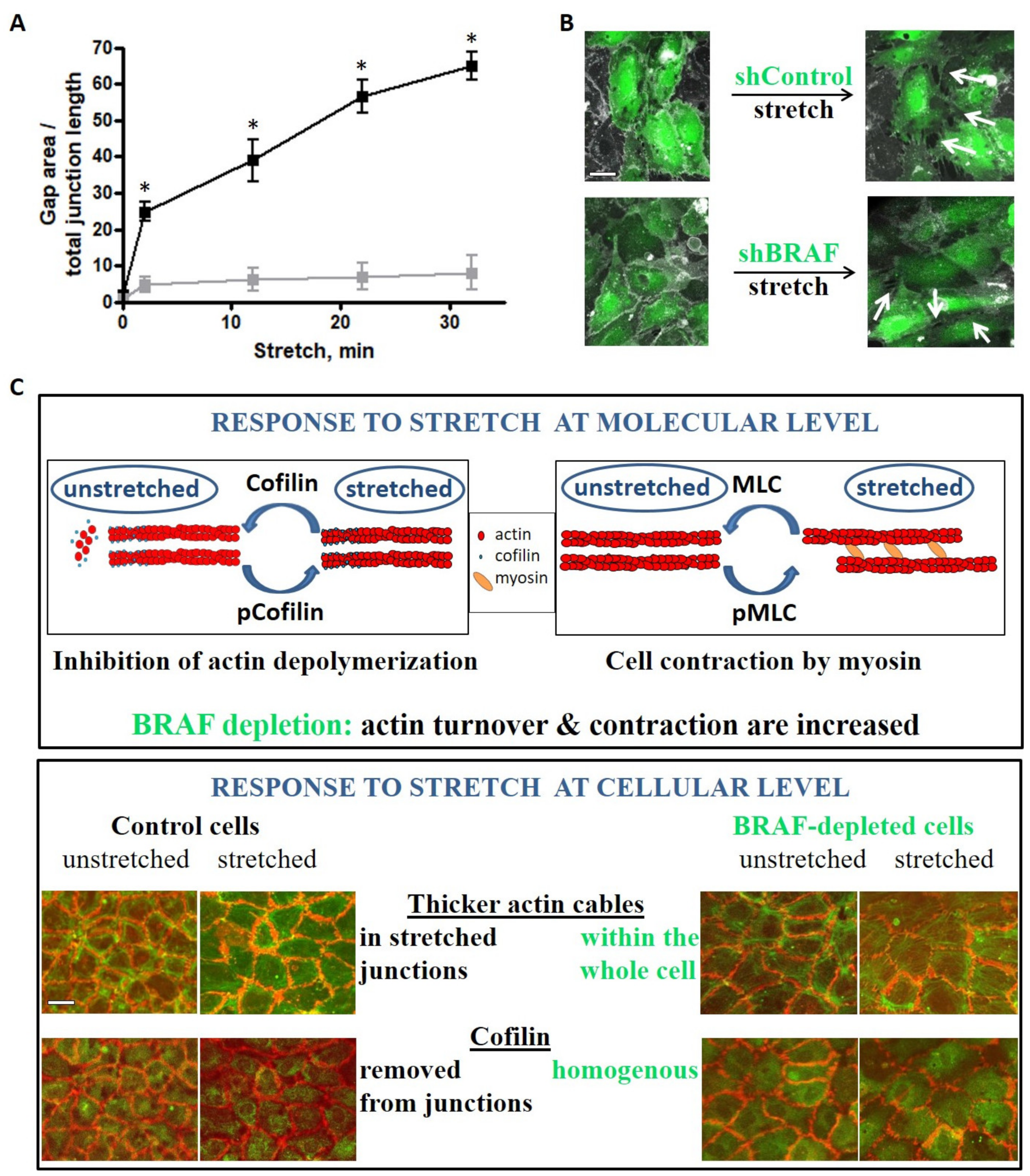
2.4. The Stretch-Induced Response of the Actin Cytoskeleton Results in a More Efficient Reinforcement of Cell–Cell Junctions through Excess Stress Fiber Formation in the Absence of BRAF
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cultured Cells/Cell Lines
4.3. Quantifications
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Strilic, B.; Offermanns, S. Intravascular survival and extravasation of tumor cells. Cancer Cell 2017, 32, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Dorard, C.; Cseh, B.; Ehrenreiter, K.; Wimmer, R.; Varga, A.; Hirschmugl, T.; Maier, B.; Kramer, K.; Furlinger, S.; Doma, E.; et al. RAF dimers control vascular permeability and cytoskeletal rearrangements at endothelial cell-cell junctions. FEBS J. 2019, 286, 2277–2294. [Google Scholar] [CrossRef] [Green Version]
- Dorland, Y.L.; Huveneers, S. Cell-cell junctional mechanotransduction in endothelial remodeling. Cell. Mol. Life Sci. 2017, 74, 279–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldenburg, J.; de Rooij, J. Mechanical control of the endothelial barrier. Cell Tissue Res. 2014, 355, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, R.; Cseh, B.; Maier, B.; Scherrer, K.; Baccarini, M. Angiogenic sprouting requires the fine tuning of endothelial cell cohesion by the Raf-1/Rok-alpha complex. Dev. Cell 2012, 22, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Varga, A.; Ehrenreiter, K.; Aschenbrenner, B.; Kocieniewski, P.; Kochanczyk, M.; Lipniacki, T.; Baccarini, M. RAF1/BRAF dimerization integrates the signal from RAS to ERK and ROKalpha. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The function of rho-associated kinases ROCK1 and ROCK2 in the pathogenesis of cardiovascular disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Newell-Litwa, K.A.; Horwitz, R.; Lamers, M.L. Non-muscle myosin II in disease: Mechanisms and therapeutic opportunities. Dis. Model. Mech. 2015, 8, 1495–1515. [Google Scholar] [CrossRef] [Green Version]
- Schnoor, M.; Garcia Ponce, A.; Vadillo, E.; Pelayo, R.; Rossaint, J.; Zarbock, A. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell. Mol. Life Sci. 2017, 74, 1985–1997. [Google Scholar] [CrossRef]
- Sumi, T.; Matsumoto, K.; Takai, Y.; Nakamura, T. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 1999, 147, 1519–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannekoek, W.J.; de Rooij, J.; Gloerich, M. Force transduction by cadherin adhesions in morphogenesis. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Charras, G.; Yap, A.S. Tensile forces and mechanotransduction at cell-cell junctions. Curr. Biol. 2018, 28, R445–R457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyeda, T.Q.; Iwadate, Y.; Umeki, N.; Nagasaki, A.; Yumura, S. Stretching actin filaments within cells enhances their affinity for the myosin II motor domain. PLoS ONE 2011, 6, e26200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laevsky, G.; Knecht, D.A. Cross-linking of actin filaments by myosin II is a major contributor to cortical integrity and cell motility in restrictive environments. J. Cell Sci. 2003, 116, 3761–3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Tatsumi, H.; Sokabe, M. Actin filaments function as a tension sensor by tension-dependent binding of cofilin to the filament. J. Cell Biol. 2011, 195, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Gordon, E.; Schimmel, L.; Frye, M. The importance of mechanical forces for in vitro endothelial cell biology. Front. Physiol. 2020, 11, 684. [Google Scholar] [CrossRef]
- Jufri, N.F.; Mohamedali, A.; Avolio, A.; Baker, M.S. Mechanical stretch: Physiological and pathological implications for human vascular endothelial cells. Vasc. Cell 2015, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, N.E.; Schildmeyer, L.A.; Bosworth, K.A.; Sakurai, Y.; Eskin, S.G.; Hurley, L.H.; McIntire, L.V. Modulating the functional contributions of c-Myc to the human endothelial cell cyclic strain response. J. Vasc. Res. 2010, 47, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Lessey, E.C.; Guilluy, C.; Burridge, K. From mechanical force to RhoA activation. Biochemistry 2012, 51, 7420–7432. [Google Scholar] [CrossRef]
- McDowell, S.A.C.; Quail, D.F. Immunological regulation of vascular inflammation during cancer metastasis. Front. Immunol. 2019, 10, 1984. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, M.; Bohec, P.; Charras, G. Mechanics of the cellular actin cortex: From signalling to shape change. Curr. Opin. Cell Biol. 2020, 66, 69–78. [Google Scholar] [CrossRef]
- Lin, T.; Zeng, L.; Liu, Y.; DeFea, K.; Schwartz, M.A.; Chien, S.; Shyy, J.Y. Rho-ROCK-LIMK-cofilin pathway regulates shear stress activation of sterol regulatory element binding proteins. Circ. Res. 2003, 92, 1296–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukov, K.G.; Jacobson, J.R.; Flores, A.A.; Ye, S.Q.; Birukova, A.A.; Verin, A.D.; Garcia, J.G. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2003, 285, L785–L797. [Google Scholar] [CrossRef] [Green Version]
- Julian, L.; Olson, M.F. Rho-associated coiled-coil containing kinases (ROCK): Structure, regulation, and functions. Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, Z.; Chen, Y.; Li, S.; Jiang, Y.; Yang, H.; Wu, C.; You, F.; Zheng, C.; Zhu, J.; et al. ROCK isoforms differentially modulate cancer cell motility by mechanosensing the substrate stiffness. Acta Biomater. 2019, 88, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Duda, M.; Kirkland, N.J.; Khalilgharibi, N.; Tozluoglu, M.; Yuen, A.C.; Carpi, N.; Bove, A.; Piel, M.; Charras, G.; Baum, B.; et al. Polarization of myosin II refines tissue material properties to buffer mechanical stress. Dev. Cell 2019, 48, 245–260.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kumar, S. Cofilin is required for polarization of tension in stress fiber networks during migration. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Hino, N.; Rossetti, L.; Marin-Llaurado, A.; Aoki, K.; Trepat, X.; Matsuda, M.; Hirashima, T. ERK-mediated mechanochemical waves direct collective cell polarization. Dev. Cell 2020, 53, 646–660.e8. [Google Scholar] [CrossRef]
- Higashi, T.; Chiba, H. Molecular organization, regulation and function of tricellular junctions. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183143. [Google Scholar] [CrossRef]
- Acharya, B.R.; Nestor-Bergmann, A.; Liang, X.; Gupta, S.; Duszyc, K.; Gauquelin, E.; Gomez, G.A.; Budnar, S.; Marcq, P.; Jensen, O.E.; et al. A Mechanosensitive RhoA pathway that protects epithelia against acute tensile stress. Dev. Cell 2018, 47, 439–452.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano, J.; Chen, M.B.; Moeendarbary, E.; Cao, X.; Shenoy, V.; Garcia-Aznar, J.M.; Kamm, R.D.; Spill, F. Balance of mechanical forces drives endothelial gap formation and may facilitate cancer and immune-cell extravasation. PLoS Comput. Biol. 2019, 15, e1006395. [Google Scholar] [CrossRef] [Green Version]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- Fehring, V.; Schaeper, U.; Ahrens, K.; Santel, A.; Keil, O.; Eisermann, M.; Giese, K.; Kaufmann, J. Delivery of therapeutic siRNA to the lung endothelium via novel Lipoplex formulation DACC. Mol. Ther. 2014, 22, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Khan, O.F.; Kowalski, P.S.; Doloff, J.C.; Tsosie, J.K.; Bakthavatchalu, V.; Winn, C.B.; Haupt, J.; Jamiel, M.; Langer, R.; Anderson, D.G. Endothelial siRNA delivery in nonhuman primates using ionizable low-molecular weight polymeric nanoparticles. Sci. Adv. 2018, 4, eaar8409. [Google Scholar] [CrossRef] [Green Version]
- Goto, C.; Nishioka, K.; Umemura, T.; Jitsuiki, D.; Sakagutchi, A.; Kawamura, M.; Chayama, K.; Yoshizumi, M.; Higashi, Y. Acute moderate-intensity exercise induces vasodilation through an increase in nitric oxide bioavailiability in humans. Am. J. Hypertens. 2007, 20, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Chatchavalvanich, S.; Rios, A.; Kawkitinarong, K.; Garcia, J.G.; Birukov, K.G. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am. J. Pathol. 2006, 168, 1749–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuebler, W.M.; Uhlig, U.; Goldmann, T.; Schael, G.; Kerem, A.; Exner, K.; Martin, C.; Vollmer, E.; Uhlig, S. Stretch activates nitric oxide production in pulmonary vascular endothelial cells in situ. Am. J. Respir. Crit. Care Med. 2003, 168, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Hu, Z.; Han, X.; Jiang, B.; Zhang, R.; Zhang, X.; Lu, Y.; Geng, C.; Li, W.; He, Y.; et al. Hypertensive stretch regulates endothelial exocytosis of Weibel-Palade bodies through VEGF receptor 2 signaling pathways. Cell Res. 2013, 23, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Caliskan, E.; de Souza, D.R.; Boning, A.; Liakopoulos, O.J.; Choi, Y.H.; Pepper, J.; Gibson, C.M.; Perrault, L.P.; Wolf, R.K.; Kim, K.B.; et al. Saphenous vein grafts in contemporary coronary artery bypass graft surgery. Nat. Rev. Cardiol. 2020, 17, 155–169. [Google Scholar] [CrossRef]
- Conte, M.S.; Mann, M.J.; Simosa, H.F.; Rhynhart, K.K.; Mulligan, R.C. Genetic interventions for vein bypass graft disease: A review. J. Vasc. Surg. 2002, 36, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.; Ramelet, A.A. Pharmacological treatment of primary chronic venous disease: Rationale, results and unanswered questions. Eur. J. Vasc. Endovasc. Surg. 2011, 41, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huveneers, S.; Oldenburg, J.; Spanjaard, E.; van der Krogt, G.; Grigoriev, I.; Akhmanova, A.; Rehmann, H.; de Rooij, J. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 2012, 196, 641–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelossof, R.; Fairchild, L.; Huang, C.H.; Widmer, C.; Sreedharan, V.T.; Sinha, N.; Lai, D.Y.; Guan, Y.; Premsrirut, P.K.; Tschaharganeh, D.F.; et al. Prediction of potent shRNAs with a sequential classification algorithm. Nat. Biotechnol. 2017, 35, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Fellmann, C.; Hoffmann, T.; Sridhar, V.; Hopfgartner, B.; Muhar, M.; Roth, M.; Lai, D.Y.; Barbosa, I.A.; Kwon, J.S.; Guan, Y.; et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013, 5, 1704–1713. [Google Scholar] [CrossRef] [Green Version]
- Aricescu, A.R.; Lu, W.; Jones, E.Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1243–1250. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollósi, A.; Pászty, K.; Kellermayer, M.; Charras, G.; Varga, A. BRAF Modulates Stretch-Induced Intercellular Gap Formation through Localized Actin Reorganization. Int. J. Mol. Sci. 2021, 22, 8989. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168989
Hollósi A, Pászty K, Kellermayer M, Charras G, Varga A. BRAF Modulates Stretch-Induced Intercellular Gap Formation through Localized Actin Reorganization. International Journal of Molecular Sciences. 2021; 22(16):8989. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168989
Chicago/Turabian StyleHollósi, Anna, Katalin Pászty, Miklós Kellermayer, Guillaume Charras, and Andrea Varga. 2021. "BRAF Modulates Stretch-Induced Intercellular Gap Formation through Localized Actin Reorganization" International Journal of Molecular Sciences 22, no. 16: 8989. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168989