
MST3 Involvement in Na+ and K+ Homeostasis with Increasing Dietary Potassium Intake


Abstract
:1. Introduction
2. Results
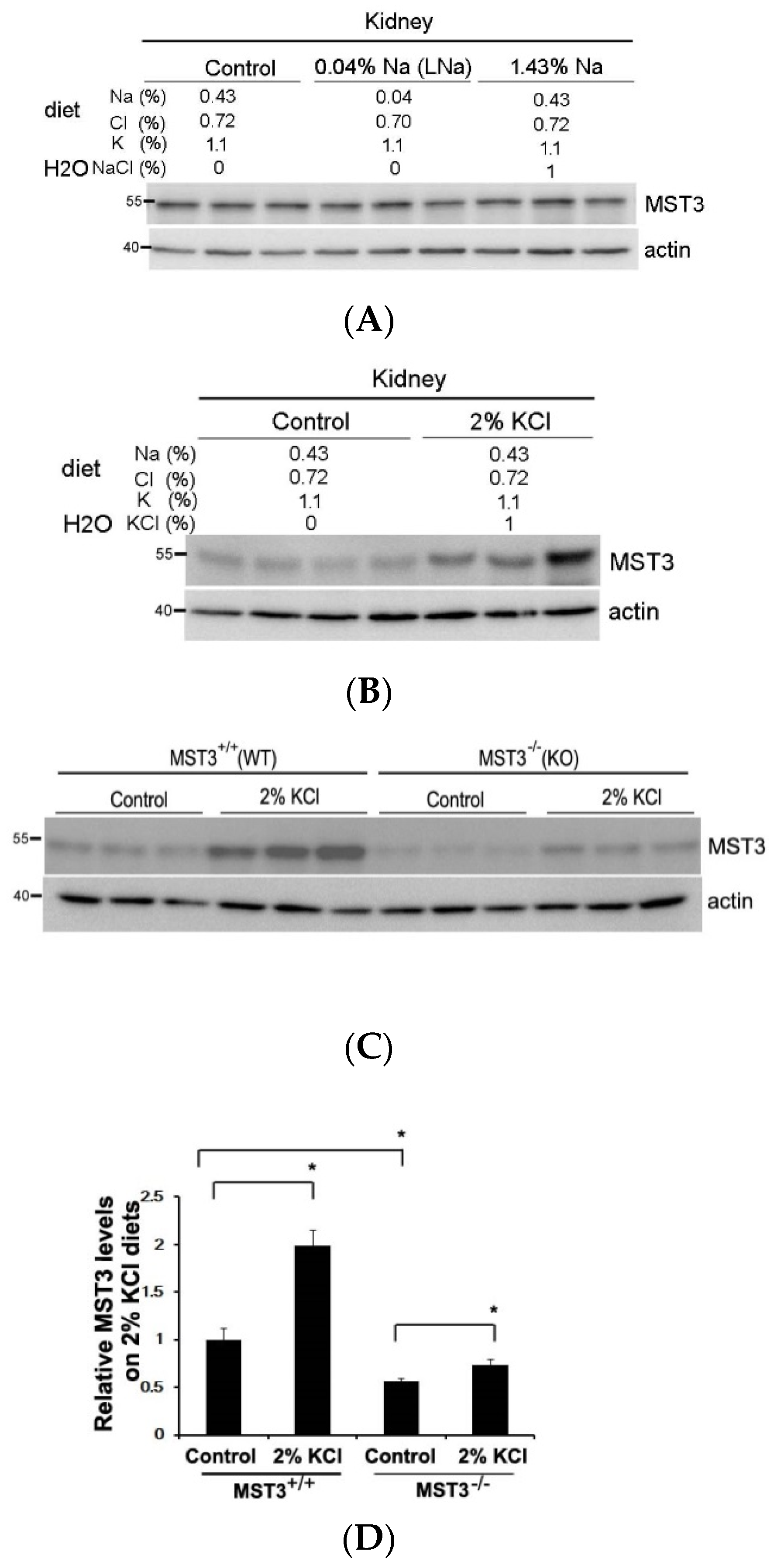
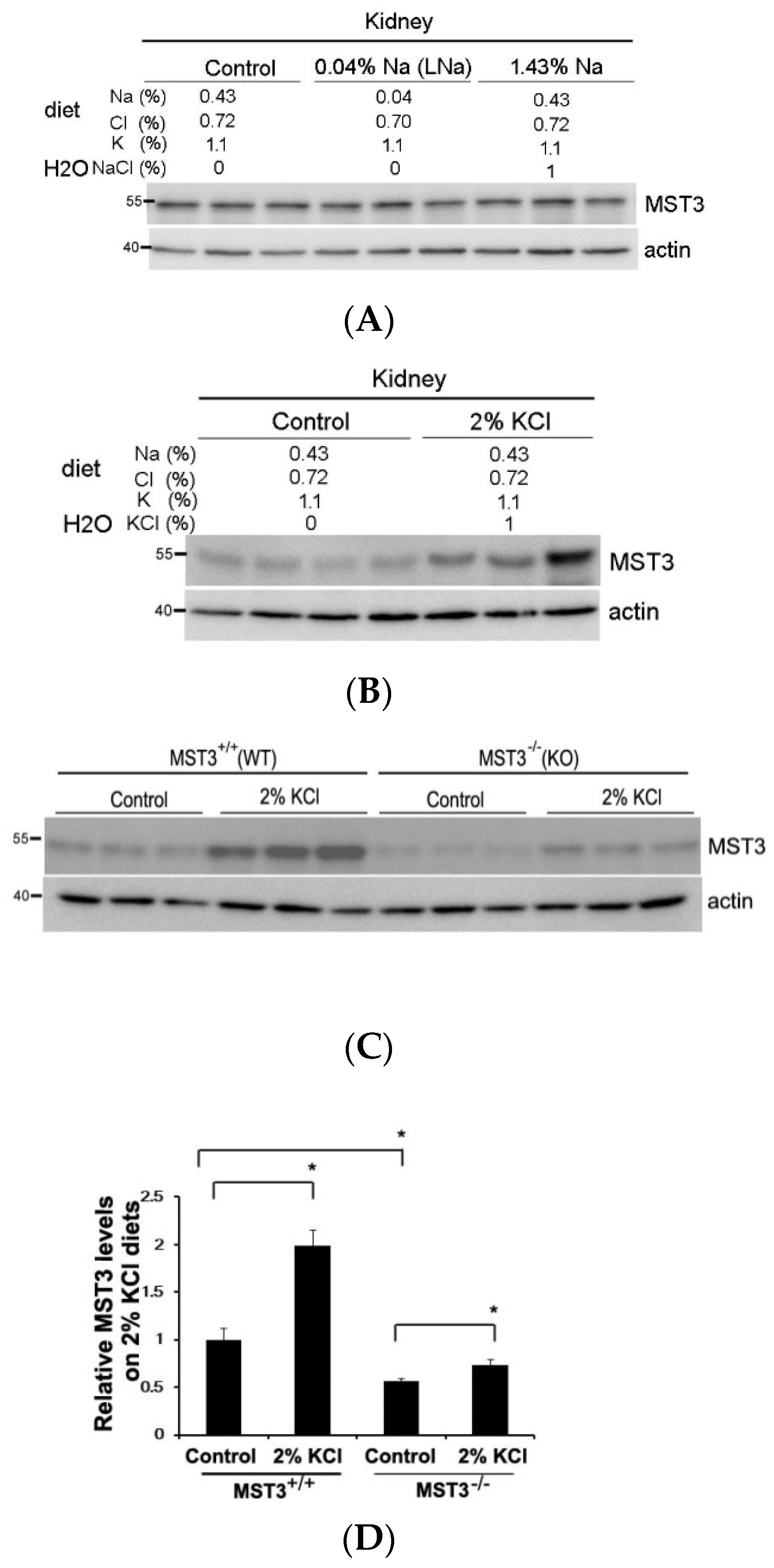
2.1. An Increase in MST3 Levels in Mouse Kidneys with Increasing K+ Intake
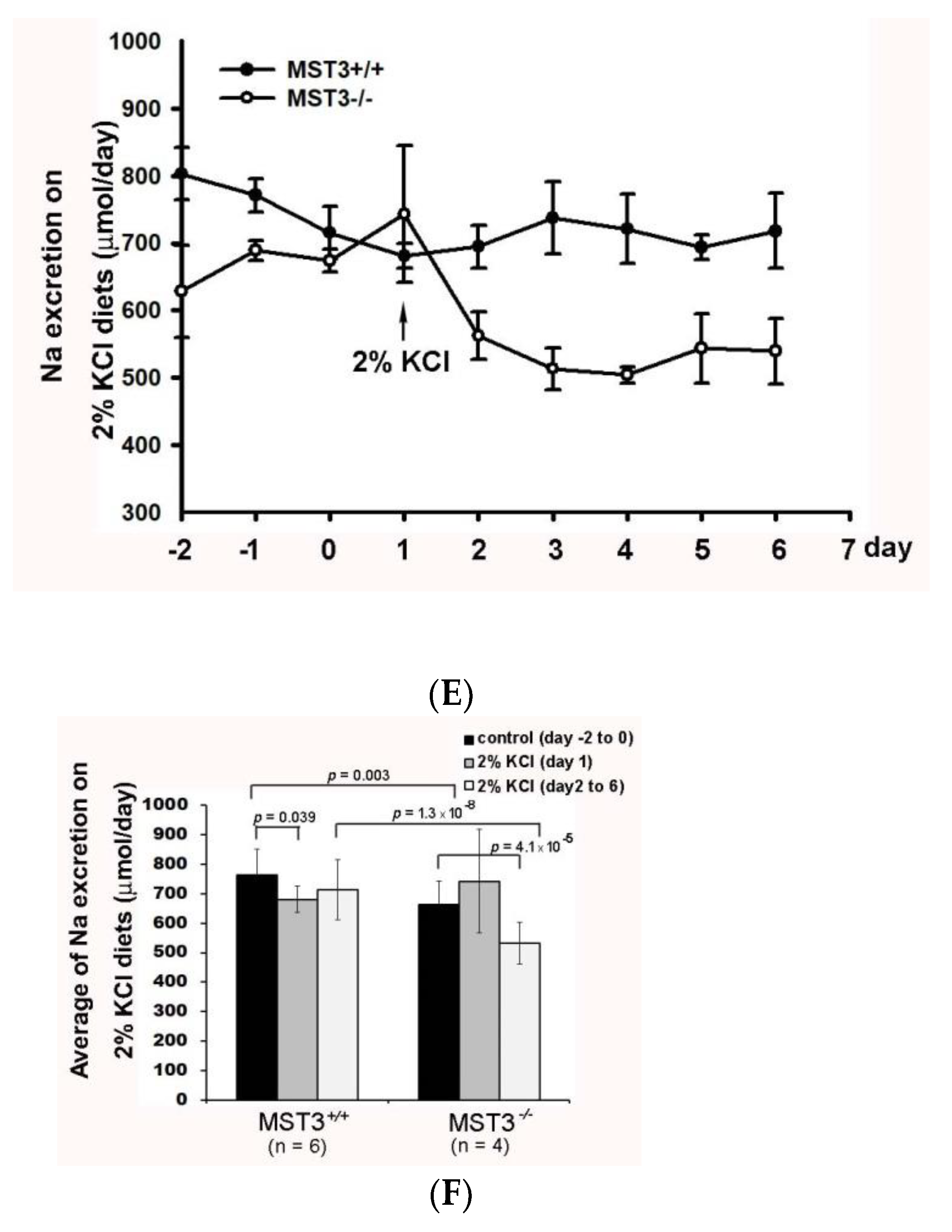
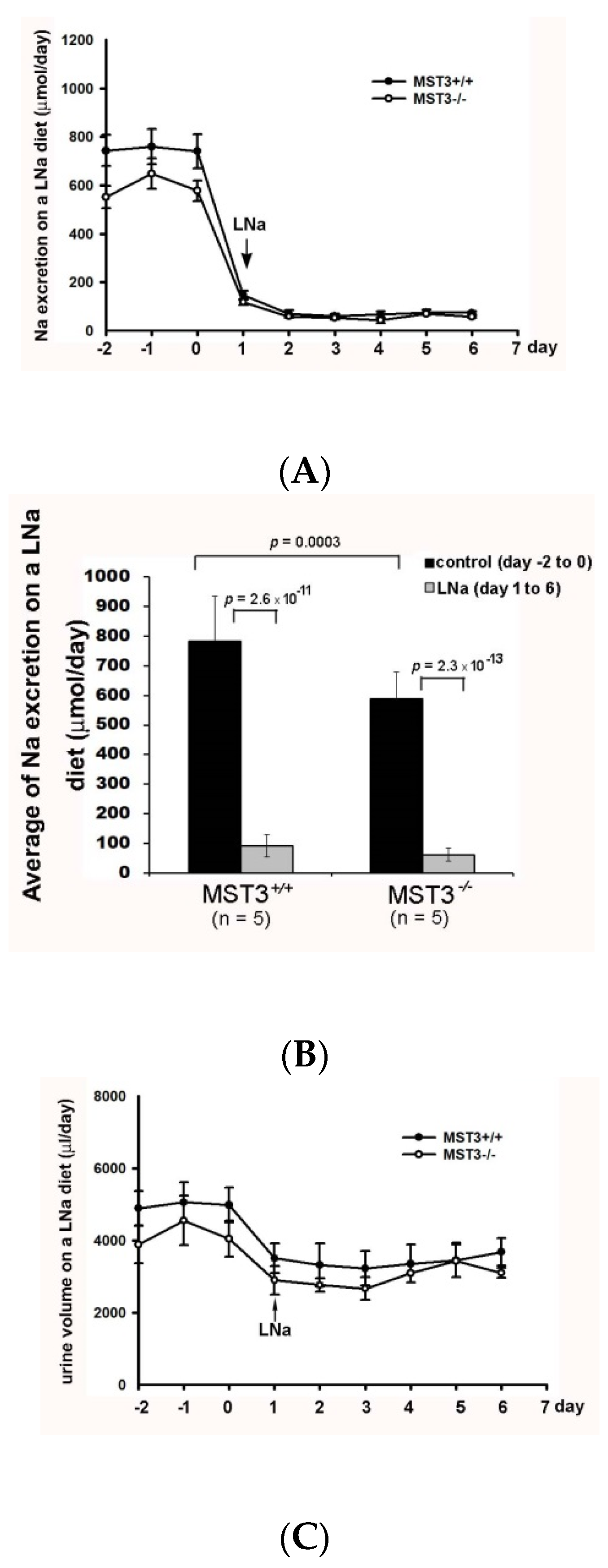
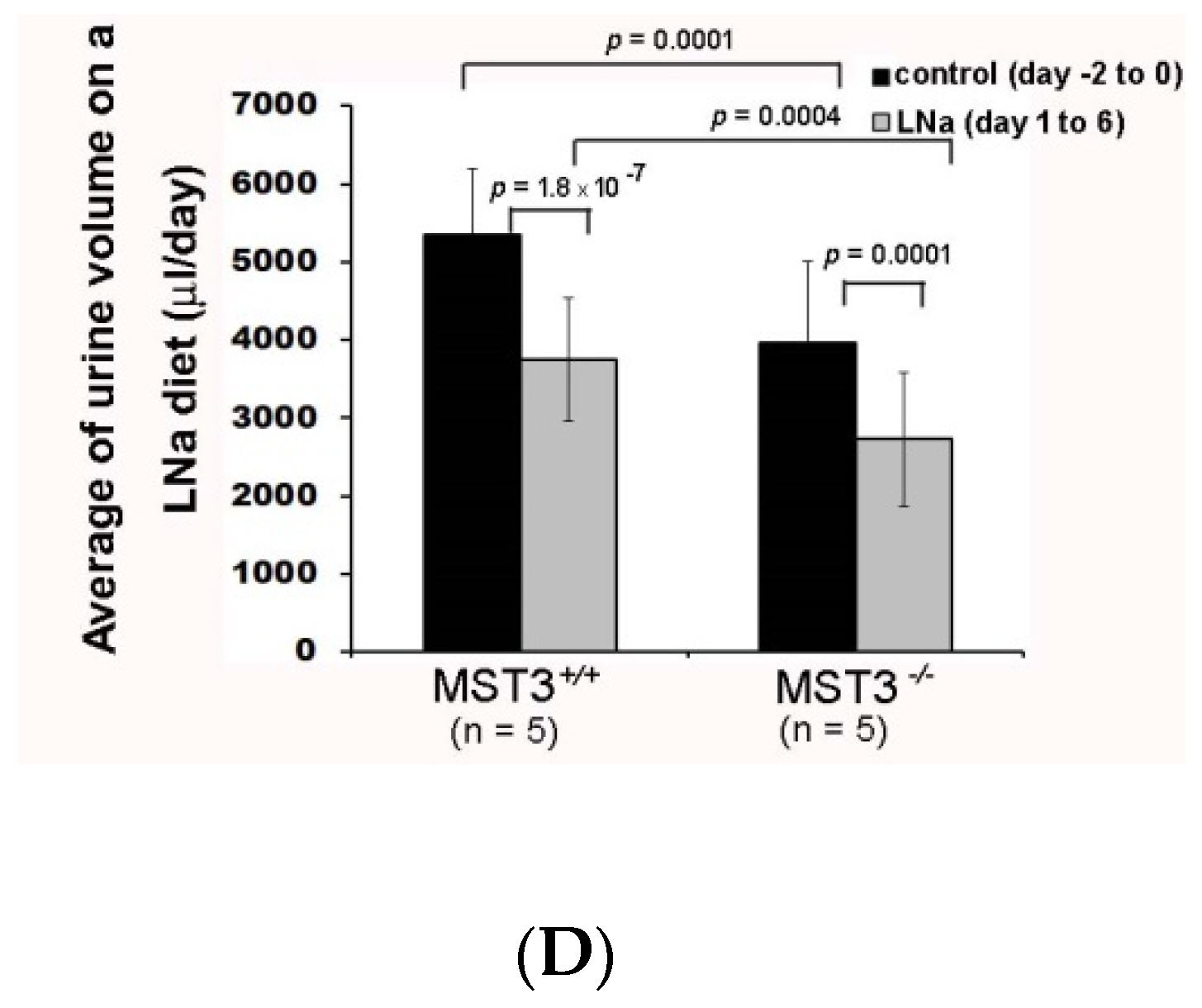
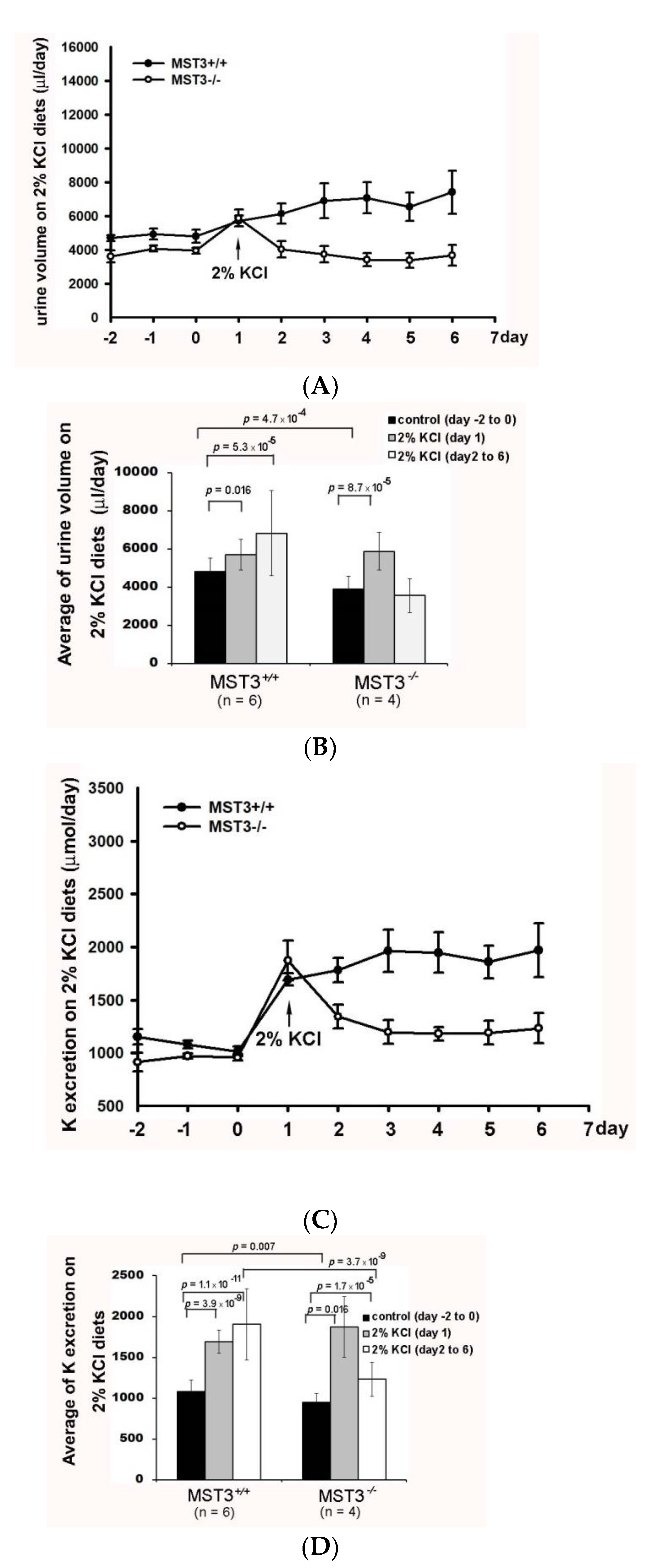
2.2. Reduction of Diuresis, Kaliuresis, and Natriuresis in MST3−/− Mice Fed 2% KCl Diets
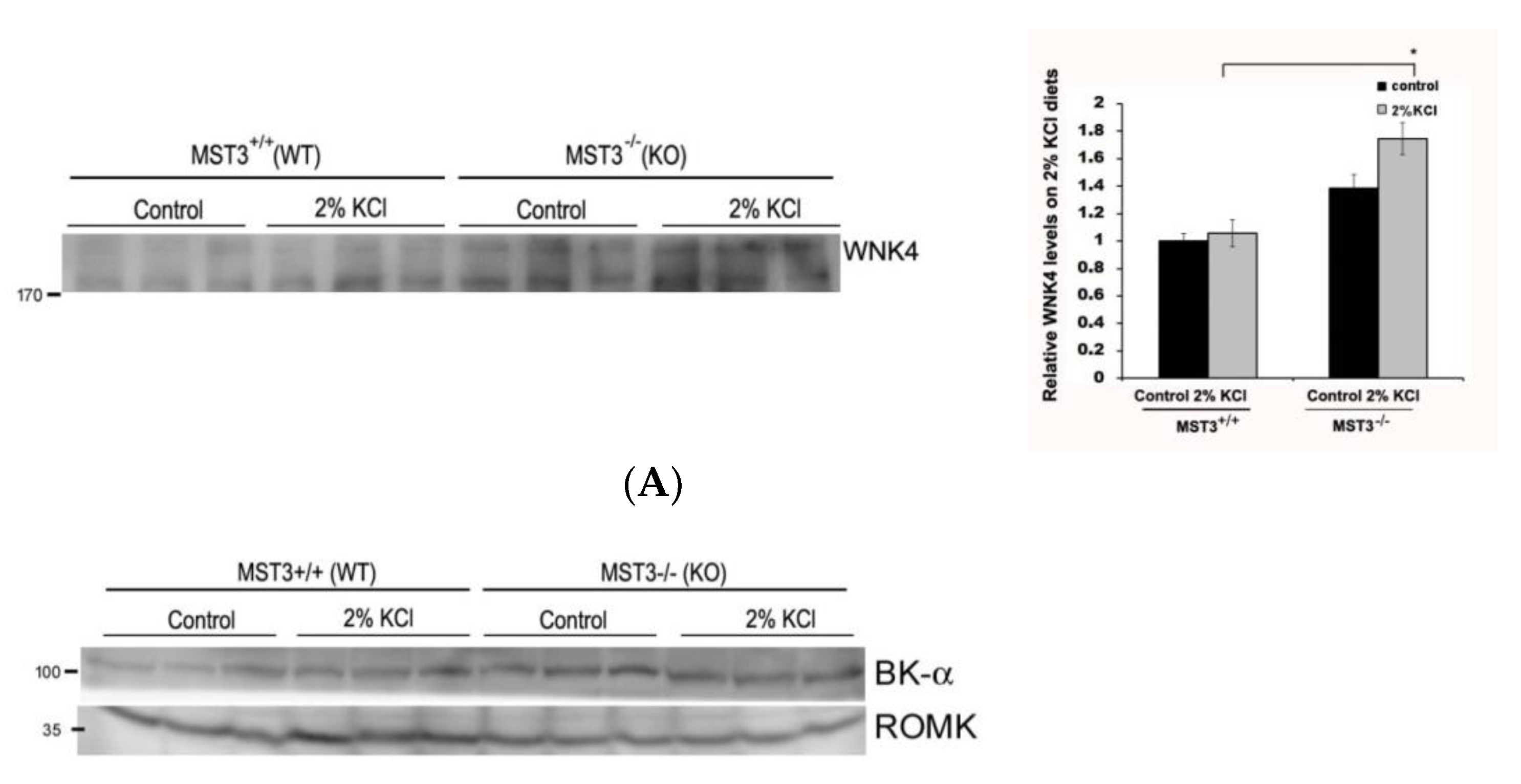
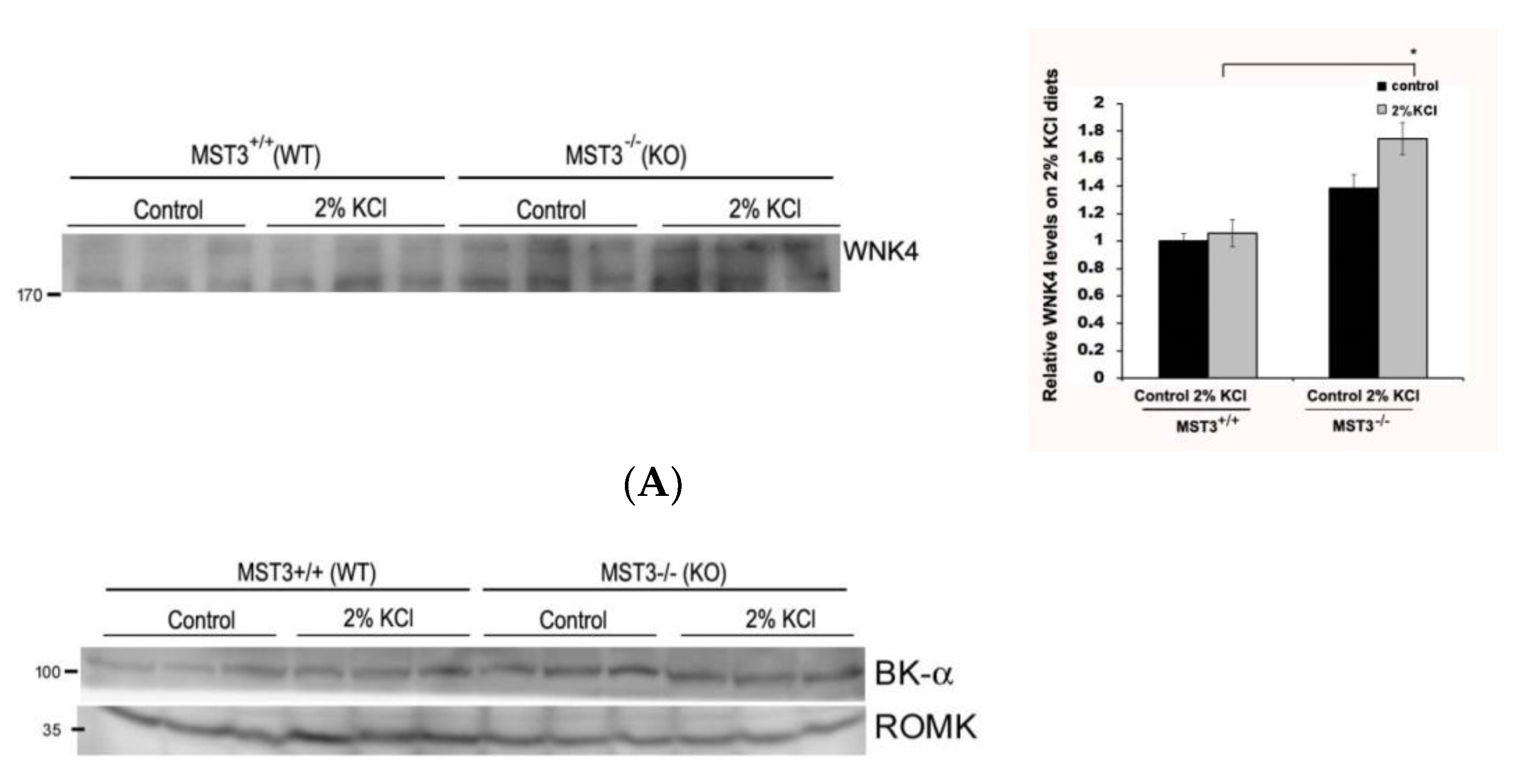
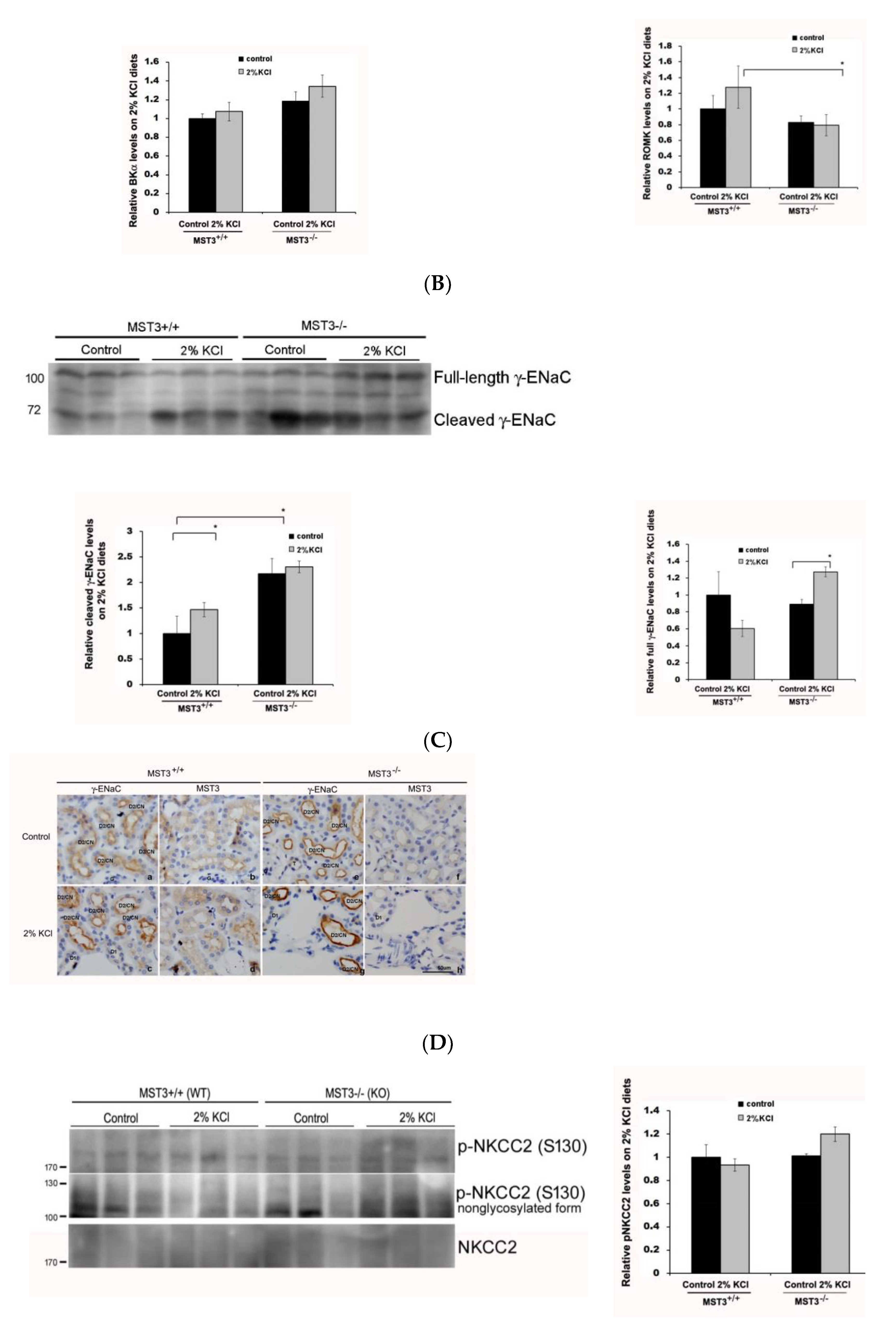
2.3. WNK4 and WNK4-Regulated Channels in 2% KCl Diet-Fed Mice
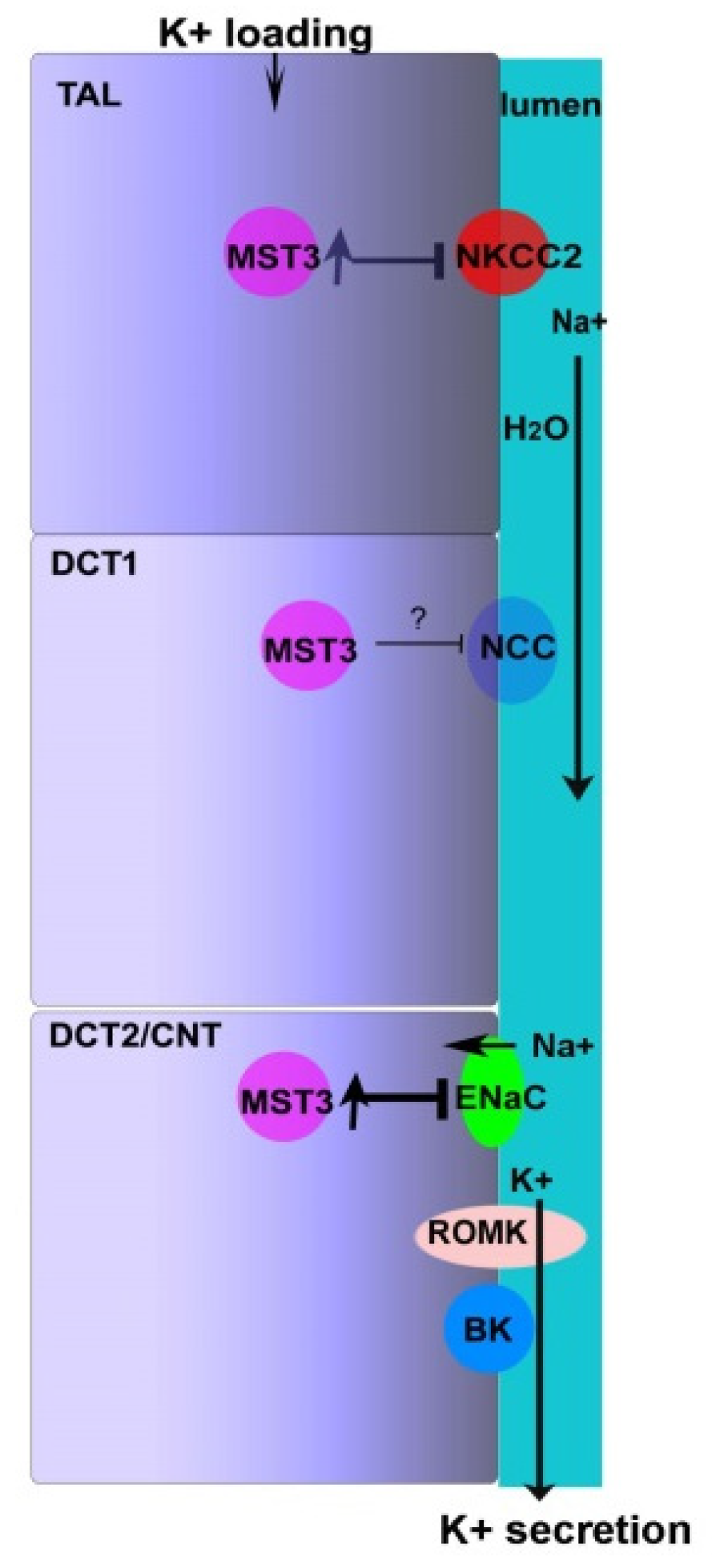
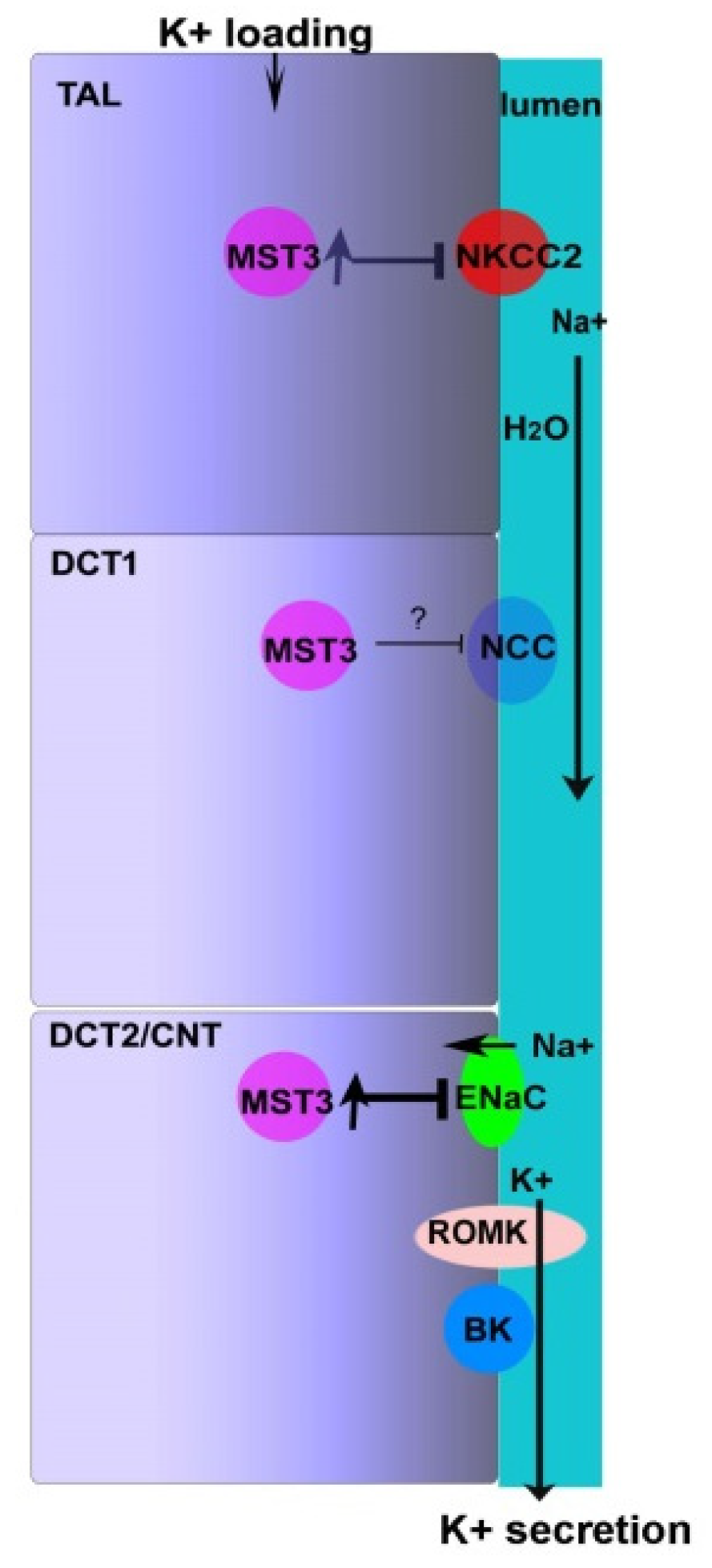
3. Discussion
4. Methods
4.1. Animals
4.2. Immunohistochemistry
4.3. Immunoblotting
4.4. Measurement of Blood Pressure, Serum Na+, and Urinary Concentrations of Na+ and K+ and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kirchner, K.A. Effect of acute potassium infusion on loop segment chloride reabsorption in the rat. Am. J. Physiol. 1983, 244, F599–F605. [Google Scholar] [CrossRef]
- Yang, L.; Xu, S.; Guo, X.; Uchida, S.; Weinstein, A.M.; Wang, T.; Palmer, L.G. Regulation of renal Na transporters in response to dietary K. Am. J. Physiol. Ren. Physiol. 2018, 315, F1032–F1041. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, M.V.; Grossmann, S.; Roesinger, M.; Gresko, N.; Todkar, A.P.; Barmettler, G.; Ziegler, U.; Odermatt, A.; Loffing-Cueni, D.; Loffing, J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 2013, 83, 811–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoda, W.; Nomura, N.; Ando, F.; Mori, Y.; Mori, T.; Sohara, E.; Rai, T.; Uchida, S. Calcineurin inhibitors block sodium-chloride cotransporter dephosphorylation in response to high potassium intake. Kidney Int. 2017, 91, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; et al. Human hypertension caused by mutations in WNK kinases. Science 2001, 293, 1107–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.C.; Lo, Y.F.; Lin, Y.W.; Lin, S.H.; Huang, C.L.; Cheng, C.J. WNK4 kinase is a physiological intracellular chloride sensor. Proc. Natl. Acad. Sci. USA 2019, 116, 4502–4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terker, A.S.; Zhang, C.; Erspamer, K.J.; Gamba, G.; Yang, C.-L.; Ellison, D.H. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2016, 89, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, K.-Y.; Gritter, M.; Vogt, L.; de Borst, M.H.; Rotmans, J.I.; Hoorn, E.J. Dietary potassium and the kidney: Lifesaving physiology. Clin. Kidney J. 2020, 13, 952–968. [Google Scholar] [CrossRef]
- Terker, A.S.; Castañeda-Bueno, M.; Ferdaus, M.Z.; Cornelius, R.J.; Erspamer, K.J.; Su, X.T.; Miller, L.N.; McCormick, J.A.; Wang, W.H.; Gamba, G.; et al. With no lysine kinase 4 modulates sodium potassium 2 chloride cotransporter activity in vivo. Am. J. Physiol. Ren. Physiol. 2018, 315, F781–F790. [Google Scholar] [CrossRef]
- Yang, S.S.; Lo, Y.F.; Wu, C.C.; Lin, S.W.; Yeh, C.J.; Chu, P.; Sytwu, H.K.; Uchida, S.; Sasaki, S.; Lin, S.H. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 2010, 21, 1868–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.H.; Yu, I.S.; Jiang, S.T.; Lin, S.W.; Chu, P.; Chen, A.; Sytwu, H.K.; Sohara, E.; Uchida, S.; Sasaki, S.; et al. Impaired phosphorylation of Na(+)-K(+)-2Cl(-) cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 17538–17543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiqi, F.H.; Zuber, A.M.; Glover, M.; Richardson, C.; Fleming, S.; Jovanovic, S.; Jovanovic, A.; O’Shaughnessy, K.M.; Alessi, D.R. Role of the WNK-activated SPAK kinase in regulating blood pressure. Embo Mol. Med. 2010, 2, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Sakamoto, K.; de los Heros, P.; Deak, M.; Campbell, D.G.; Prescott, A.R.; Alessi, D.R. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J. Cell Sci. 2011, 124, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Kleta, R.; Bockenhauer, D. Salt-Losing Tubulopathies in Children: What’s New, What’s Controversial? J. Am. Soc. Nephrol. 2018, 29, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalla, V.; Hallows, K.R. Mechanisms of ENaC regulation and clinical implications. J. Am. Soc. Nephrol. 2008, 19, 1845–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wang, L.; Su, X.T.; Zhang, J.; Lin, D.H.; Wang, W.H. ENaC and ROMK activity are inhibited in the DCT2/CNT of TgWnk4(PHAII) mice. Am. J. Physiol. Ren. Physiol. 2017, 312, F682–F688. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.J.; Kan, W.C.; Yang, S.S.; Jiang, S.T.; Wu, S.N.; Ling, P.; Bao, B.Y.; Lin, C.Y.; Yang, Z.Y.; Weng, Y.P.; et al. MST3 is involved in ENaC-mediated hypertension. Am. J. Physiol. Ren. Physiol. 2019, 317, F30–F42. [Google Scholar] [CrossRef]
- Lu, T.-J.; Chan, C.-H.; Ling, P.; Chao, Y.-M.; Bao, B.-Y.; Chiang, C.-Y.; Lee, T.-H.; Weng, Y.-P.; Kan, W.-C.; Lu, T.-L. MST3 (mammalian Ste20-like protein kinase 3), a novel gene involved in ion homeostasis and renal regulation of blood pressure in spontaneous hypertensive rats. Int. Urol. Nephrol. 2018, 50, 2299–2307. [Google Scholar] [CrossRef]
- Rengarajan, S.; Lee, D.H.; Oh, Y.T.; Delpire, E.; Youn, J.H.; McDonough, A.A. Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am. J. Physiol. Ren. Physiol. 2014, 306, F1059–F1068. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Wang, H.R.; Huang, S.K.; Huang, C.L. Intersectin links WNK kinases to endocytosis of ROMK1. J. Clin. Investig. 2007, 117, 1078–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, J.; Zhang, X.; Wang, D.; Li, J.; Zhou, B.; Shi, Z.; Gu, D.; Denson, D.D.; Eaton, D.C.; Cai, H. WNK4 kinase inhibits Maxi K channel activity by a kinase-dependent mechanism. Am. J. Physiol. Ren. Physiol. 2011, 301, F410–F419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ares, G.R.; Caceres, P.S.; Ortiz, P.A. Molecular regulation of NKCC2 in the thick ascending limb. Am. J. Physiol. Ren. Physiol. 2011, 301, F1143–F1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, L.G.; Frindt, G. Aldosterone and potassium secretion by the cortical collecting duct. Kidney Int. 2000, 57, 1324–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, B.F.; Clegg, D.J. Physiology and pathophysiology of potassium homeostasis. Adv. Physiol. Educ. 2016, 40, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, P.R.; Sansom, S.C. BK channels and a new form of hypertension. Kidney Int. 2010, 78, 956–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieg, T.; Vallon, V.; Sausbier, M.; Sausbier, U.; Kaissling, B.; Ruth, P.; Osswald, H. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int. 2007, 72, 566–573. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.; Zhang, J.; Puthumana, J.; Stone, K.L.; Lifton, R.P. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc. Natl. Acad. Sci. USA 2013, 110, 7838–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castañeda-Bueno, M.; Arroyo, J.P.; Zhang, J.; Puthumana, J.; Yarborough, O.; Shibata, S.; Rojas-Vega, L.; Gamba, G.; Rinehart, J.; Lifton, R.P. Phosphorylation by PKC and PKA regulate the kinase activity and downstream signaling of WNK4. Proc. Natl. Acad. Sci. USA 2017, 114, E879–E886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.-H.; Yue, P.; Rinehart, J.; Sun, P.; Wang, Z.; Lifton, R.; Wang, W.-H. Protein phosphatase 1 modulates the inhibitory effect of With-no-Lysine kinase 4 on ROMK channels. Am. J. Physiol. Ren. Physiol. 2012, 303, F110–F119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penton, D.; Czogalla, J.; Wengi, A.; Himmerkus, N.; Loffing-Cueni, D.; Carrel, M.; Rajaram, R.D.; Staub, O.; Bleich, M.; Schweda, F.; et al. Extracellular K(+) rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl(-) -dependent and independent mechanisms. J. Physiol. 2016, 594, 6319–6331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, W.-C.; Lu, T.-L.; Ling, P.; Lee, T.-H.; Cho, C.-Y.; Huang, C.-Y.F.; Jeng, W.-Y.; Weng, Y.-P.; Chiang, C.-Y.; Wu, J.B.; et al. Pervanadate induces Mammalian Ste20 Kinase 3 (MST3) tyrosine phosphorylation but not activation. J. Inorg. Biochem. 2016, 160, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.J.; Lai, W.Y.; Huang, C.Y.; Hsieh, W.J.; Yu, J.S.; Hsieh, Y.J.; Chang, W.T.; Leu, T.H.; Chang, W.C.; Chuang, W.J.; et al. Inhibition of cell migration by autophosphorylated mammalian sterile 20-like kinase 3 (MST3) involves paxillin and protein-tyrosine phosphatase-PEST. J. Biol. Chem. 2006, 281, 38405–38417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.J.; Huang, C.Y.; Yuan, C.J.; Lee, Y.C.; Leu, T.H.; Chang, W.C.; Lu, T.L.; Jeng, W.Y.; Lai, M.D. Zinc ion acts as a cofactor for serine/threonine kinase MST3 and has a distinct role in autophosphorylation of MST3. J. Inorg. Biochem. 2005, 99, 1306–1313. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diet | Control Diet (0.43% Na, 1.1% K) (n = 5) | High Salt (8% Na, 1.1% K) (n = 5) |
|---|---|---|
| Weight, g | 19.55 ± 2.12 | 21.94 ± 1.52 |
| Water intake, mL | 4.96 ± 0.52 | 8.62 ± 1.01 |
| Food intake, g | 3.17 ± 0.43 | 8.82 ± 1.89 |
| Value | |||
|---|---|---|---|
| 16 Days | MST3+/+ (WT) | MST3 −/− | |
| Plasma [Na+] (mmol/L) | control diet | 152.33 ± 0.52 | 153.4 ± 0.89 * |
| 2% KCl diets | 152.5 ± 0.84 | 154.67 ± 1.53 * | |
| Blood pressure (mmHg) | control diet | 117 ± 9 | 130 ± 13 * |
| 2% KCl diets | 118 ± 10 | 131 ± 9 * | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, C.-H.; Wu, S.-N.; Bao, B.-Y.; Li, H.-W.; Lu, T.-L. MST3 Involvement in Na+ and K+ Homeostasis with Increasing Dietary Potassium Intake. Int. J. Mol. Sci. 2021, 22, 999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030999
Chan C-H, Wu S-N, Bao B-Y, Li H-W, Lu T-L. MST3 Involvement in Na+ and K+ Homeostasis with Increasing Dietary Potassium Intake. International Journal of Molecular Sciences. 2021; 22(3):999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030999
Chicago/Turabian StyleChan, Chee-Hong, Sheng-Nan Wu, Bo-Ying Bao, Houng-Wei Li, and Te-Ling Lu. 2021. "MST3 Involvement in Na+ and K+ Homeostasis with Increasing Dietary Potassium Intake" International Journal of Molecular Sciences 22, no. 3: 999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030999