
Species-Specific Regulation of TRPM2 by PI(4,5)P2 via the Membrane Interfacial Cavity


Institute of Physiology, Medical Faculty, RWTH Aachen University Hospital, D52057 Aachen, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(9), 4637; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094637
Submission received: 8 March 2021
/
Revised: 21 April 2021
/
Accepted: 26 April 2021
/
Published: 28 April 2021
(This article belongs to the Special Issue TRPM Channels)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The human apoptosis channel TRPM2 is stimulated by intracellular ADR-ribose and calcium. Recent studies show pronounced species-specific activation mechanisms. Our aim was to analyse the functional effect of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), commonly referred to as PIP2, on different TRPM2 orthologues. Moreover, we wished to identify the interaction site between TRPM2 and PIP2. We demonstrate a crucial role of PIP2, in the activation of TRPM2 orthologues of man, zebrafish, and sea anemone. Utilizing inside-out patch clamp recordings of HEK-293 cells transfected with TRPM2, differential effects of PIP2 that were dependent on the species variant became apparent. While depletion of PIP2 via polylysine uniformly caused complete inactivation of TRPM2, restoration of channel activity by artificial PIP2 differed widely. Human TRPM2 was the least sensitive species variant, making it the most susceptible one for regulation by changes in intramembranous PIP2 content. Furthermore, mutations of highly conserved positively charged amino acid residues in the membrane interfacial cavity reduced the PIP2 sensitivity in all three TRPM2 orthologues to varying degrees. We conclude that the membrane interfacial cavity acts as a uniform PIP2 binding site of TRPM2, facilitating channel activation in the presence of ADPR and Ca2+ in a species-specific manner.
1. Introduction
Transient receptor potential melastatin type 2 (TRPM2) is a non-selective cation channel that belongs to the TRPM subfamily, consisting of TRPM1 to TRPM8. Extensive cryo-EM studies in the past few years have revealed the complex structure of TRPM2 (reviewed in [1]). As a result, the channel was identified as a homotetramer with each monomer consisting of an N-terminal TRP melastatin homology region (MHR) formed by four subdomains (MHR1-4). The subsequent transmembrane region (TM) is composed of the S1–S4 voltage sensor-like domain followed by the pore-forming S5–S6 domain. Right after the TM region are the TRP helices, a rib helix, and a pole helix with the adjacent NUDT9 homology (NUDT9H) domain at the C-terminus.
The principal stimulus for gating of TRPM2 channels is ADP-ribose (ADPR) which is produced in many cells during oxidative stress [2,3]. Moreover, intracellular calcium is an important co-agonist, leading to self-enhancing processes during TRPM2 activation because the channel enables calcium influx [4,5]. Therefore, TRPM2 takes part in many physiological processes such as immune response [6], oxidative stress-induced apoptosis [7], insulin secretion [8,9], and temperature regulation [10].
Recently, functional analyses of species variants as well as several cryo-EM studies have revealed elementary species-specific differences in the ADPR-dependent gating mechanism of TRPM2 (reviewed in [1]). The presence of an additional ADPR binding pocket beside the classical one in the NUDT9H domain is now experimentally confirmed [11,12,13,14,15,16]. This dualism in ADPR binding is present in all channel orthologues investigated so far, but the functional importance of the two binding pockets is quite different depending on the species variant. In TRPM2 of the sea anemone (nvTRPM2) an N-terminal ADPR binding pocket (NUDT9H) exclusively has ADPRase function [11,13,16,17]. In contrast, in hsTRPM2 and drTRPM2 the presence of the NUDT9H domain is essential for channel function whilst significant ADPRase activity is missing [3,17,18,19,20,21]. Moreover, in hsTRPM2 binding of ADPR to both binding pockets seems to be required for channel gating [14], while in drTRPM2 binding of ADPR to NUDT9H is rather unimportant [13,22]. Thus, it is not entirely clear whether the crucial role of the NUDT9H domain in vertebrate TRPM2 derives from direct ADPR binding or solely from the facilitation of interdomain interactions [13,14,15,22], that are absent in non-vertebrate orthologues [12,16,17]. Differences in how ADPR stimulates TRPM2 channels in different species are not confined to the intramolecular mechanisms of how gating is achieved but extend to the potency of ADPR. Considerably higher ADPR concentrations are required in hsTRPM2 compared with the other two orthologues [3,13,23]. Due to these species-specific gating mechanisms of TRPM2, it is of particular interest to study different TRPM2 orthologues with regard to various TRPM2 modulators to understand the full scope of channel activation.
In the past decades, a number of phospholipids, particularly phosphatidylinositol 4,5-bisphosphate (PI(4,5P2), referred to as PIP2, have been shown to regulate the activity of a vast variety of ion channels and transport proteins [24]. This observation was first reported by Choquette et al., in 1984 via a study investigating brain Ca2+-ATPases in liposomes in the presence and absence of PIP2. They found that PIP2 stimulates the activity of the Ca2+-ATPases [25]. Since then, several other ion channels were identified to be regulated by PIP2 including members of the TRP family. Whilst some channels, including TRPM4, TRPM5, TRPM8, TRPV5, and TRPV6 are positively regulated by PIP2 [26], others are negatively regulated including TRPC4, TRPP2, and TRPP3 [27,28,29]. In 2012, the human species variant of TRPM2 was first shown to be positively regulated by PIP2 when expressed in Xenopus laevis oocytes, where PIP2 was proposed to regulate the Ca2+ sensitivity of TRPM2 activation [30]. This positive regulation by PIP2 is also present in nvTRPM2 [31]. While the PIP2-interaction sites for TRPM2 have not yet been identified, they are discovered for their closest relative within the TRP family, TRPM8. In TRPM8, mutation of conserved positively charged amino acids within the membrane interfacial cavity reduces the sensitivity of the channel for PIP2 and increases inhibition by PIP2 depletion [27,32,33]. The membrane interfacial cavity is formed by the pre-S1 domain, the junction between S4 and S5, TRP domain, and MHR4 from the adjacent subunit. Since these domains and residues are highly conserved within the TRPM family and similar mutations in TRPM5 result in reduced PIP2 induced currents, it is thought that PIP2 interaction with TRPM2 is facilitated in a similar fashion.
Therefore, the aim of the study was a comprehensive analysis of the effect of PIP2 on TRPM2 activity. Specifically, we focused on the PIP2 binding site in TRPM2 and potential species-dependent differences due to the unique species-specific gating mechanism. These experiments should lead to a better understanding of how TRPM2 activity is modulated by PIP2 and may provide a new aspect of TRPM2 regulation in physiological processes.
Here, we show that three species variants of TRPM2, representing a broad spectrum of metazoan evolution, are positively regulated by PIP2 when expressed in HEK 293 cells. The comparative analysis shows, however, that the extent of the positive regulation by PIP2 differs depending on the species variant. In addition, this study provides the first experimental evidence for a uniform interaction site between TRPM2 and PIP2 which is similar to the membrane interfacial cavity previously described for TRPM8 [32].
2. Results
2.1. PIP2 Is a Necessary Cofactor for TRPM2 Activation in Mammalian Cells
To study the effects of PIP2 on TRPM2 channels, inside-out patch clamp experiments were performed on different species variants of TRPM2 heterologously expressed in HEK 293 cells: Human TRPM2 (hsTRPM2), zebrafish (Danio rerio) TRPM2 (drTRPM2), and sea anemone (Nematostella vectensis) TRPM2 (nvTRPM2). Expression was so high that macroscopic currents rather than single-channel events were consistently observed after stimulation. PIP2 and other PIP analogues were used in their synthetic diC8-form which are commercially available and sufficiently water-soluble and applied to the cytosolic side of the membrane of inside-out patches by a multi-barrel perfusion system.
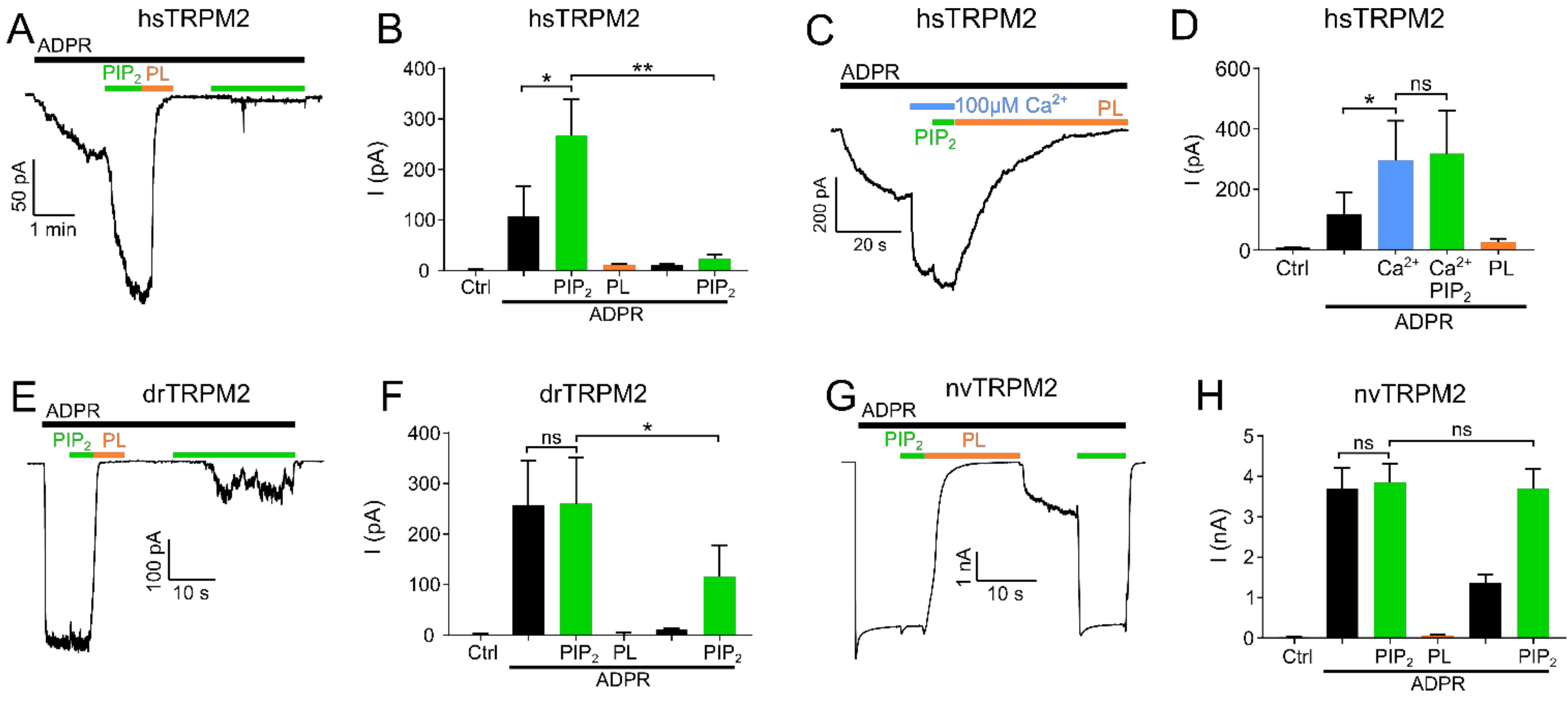
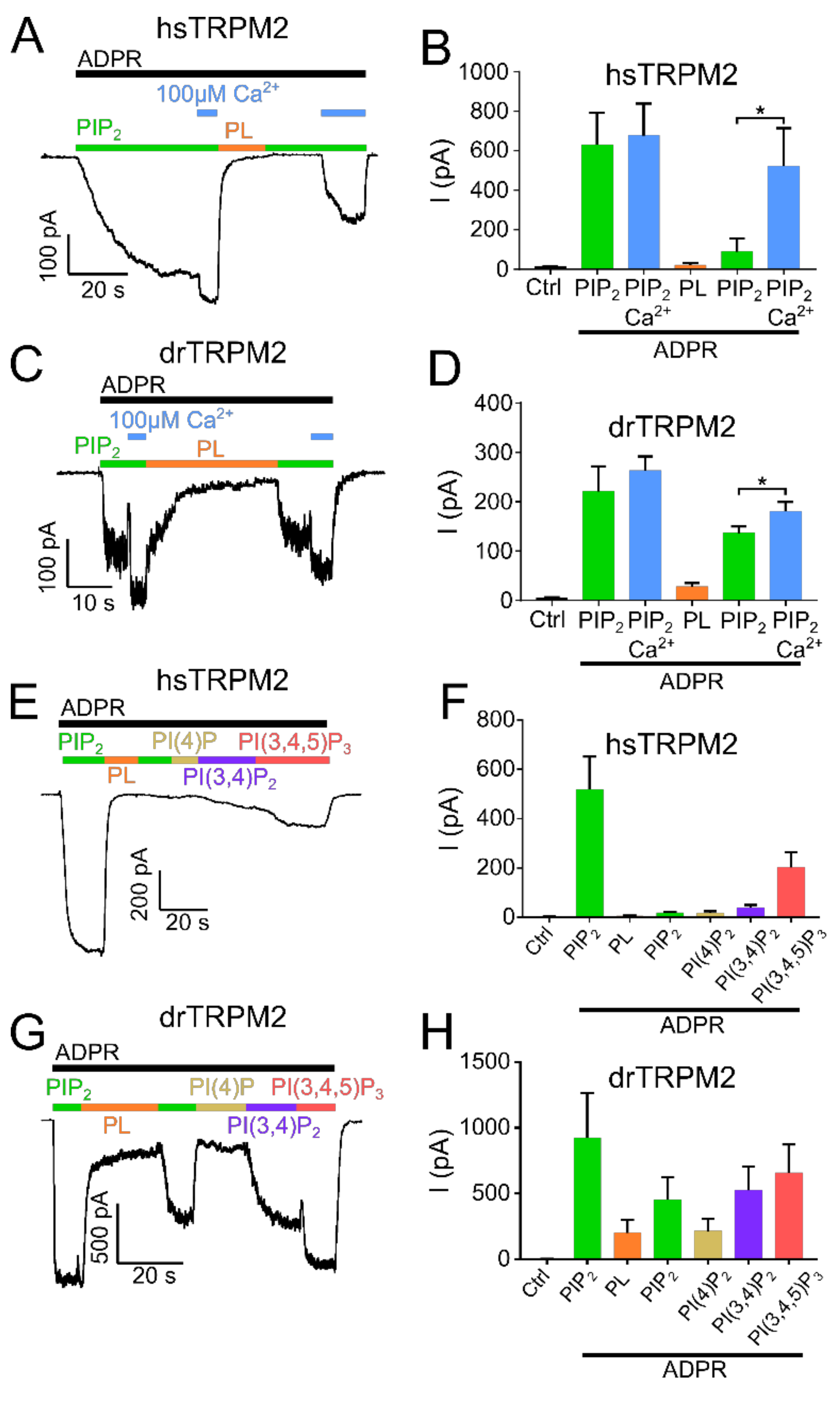
In hsTRPM2, no currents were observed in the absence of ADPR but the continuous application of ADPR (300 µM) resulted in gradually developing, long-lasting macroscopic currents (Figure 1A,B). When a plateau was reached after approximately 2 min, PIP2 (25 µM) was applied in addition to ADPR. This increased the current significantly by about 2.5-fold. Interestingly, the strongly increasing intracellular concentration of Ca2+ (100 µM) prior to PIP2 application caused a significant current increase that was only marginally further increased by the addition of 25 µM PIP2 (Figure 1C,D). This result shows that when the applied concentrations of Ca2+ are too low, the starting open probability of hsTRPM2 is also low, and therefore can be further increased by PIP2 application. Following PIP2 addition, a subsequent switch to a perfusion solution without PIP2 but with ADPR and polylysine (15 µg/mL) completely and rapidly abolished the currents. Polylysine acts as a PIP2 scavenger and therefore inhibits the activity of PIP2 activated ion channels [34,35]. Washout of polylysine, under the continuing application of ADPR, failed to restore any currents. The current rescue was also not achieved with the subsequent application of PIP2 (25 µM), inducing either negligible or no response (Figure 1A,B) even with PIP2 concentrations up to 100 µM (Supplementary Figure S1A). Only when Ca2+ (100 µM) was supplemented to PIP2, a partial rescue of the current was achieved (Figure 2A,B). This current restoration was PIP2 and calcium-dependent as high calcium concentrations in the absence of PIP2 were not sufficient to restore currents (Supplementary Figure S1C,D). We next tested whether other PI derivatives, PI(4)P, PI(3,4)P2, and PI(3,4,5)P3, might be more effective than PIP2 on the restoration of polylysine-abolished currents. Only for PI(3,4,5)P3 was such a tendency observed (Figure 2E,F). As hsTRPM2 and drTRPM2 currents also show rundown in HEK-293 cells (Supplementary Figure S2B,C), although in a strongly delayed manner, if compared to the results obtained from Xenopus oocytes [17,30] while nvTRPM2 currents do not show any rundown at all (Supplementary Figure S2D), current restoration upon polylysine treatment could be affected by this process. However, over the time course of our recordings rundown should only marginally affect current restoration.
Examining drTRPM2, a concentration of 100 µM ADPR (please refer to Supplementary Figure S2A for ADPR dose-response recordings of hs-, dr- and nvTRPM2) was sufficient to evoke strong and rapid increases in currents but the additional application of PIP2 did not increase the ADPR-induced current further, suggesting that drTRPM2 may already be saturated with the pre-existing PIP2 concentration in the patch (Figure 1E,F). In addition, when compared to hsTRPM2 starting open probability of drTRPM2 at 1 µM calcium may be already close to the maximum, resulting in no further current increase during PIP2 application. Similar to hsTRPM2, polylysine caused a complete current decay. Subsequent washout of polylysine only led to minuscule currents whereas the additional application of PIP2 rescued about half of the initial current amplitudes (Figure 1E,F). In addition, increasing the Ca2+ concentration to 100 µM significantly increases current restoration via PIP2 (Figure 2C,D), even though this effect is less pronounced compared with hsTRPM2. In contrast to hsTRPM2, minor currents were restored via high calcium concentrations (100 µM) in absence of PIP2 (Supplementary Figure S1E,F). Additionally, application of the PIP2 derivatives PI(3,4)P2 and PI(3,4,5)P3 tends to improve current restoration while PI(4)P has no effect (Figure 2G,H).
In nvTRPM2, ADPR (100 µM) caused a rapid current increase which was not further enhanced by PIP2 (Figure 1G,H). Again, similar to drTRPM2 this may be caused by an increased starting open probability at 1 µM Ca2+ compared with hsTRPM2. Once more, polylysine caused the current to decay completely. In contrast to the findings on the other two orthologues, washout of polylysine led to a moderate current increase, and the current was completely restored to the initial maxima by the second addition of PIP2. Therefore, nvTRPM2 is the only TRPM2 species tested in this study that was able to fully recover the initial current via PIP2 after polylysine was used to scavenge PIP2. Our data suggest that even though all three TRPM2 species variants are positively regulated via PIP2, there are significant differences in the way PIP2 addition or withdrawal affects channel activity in the different species variants.
2.2. PIP2 Sensitivity Varies among TRPM2 Species
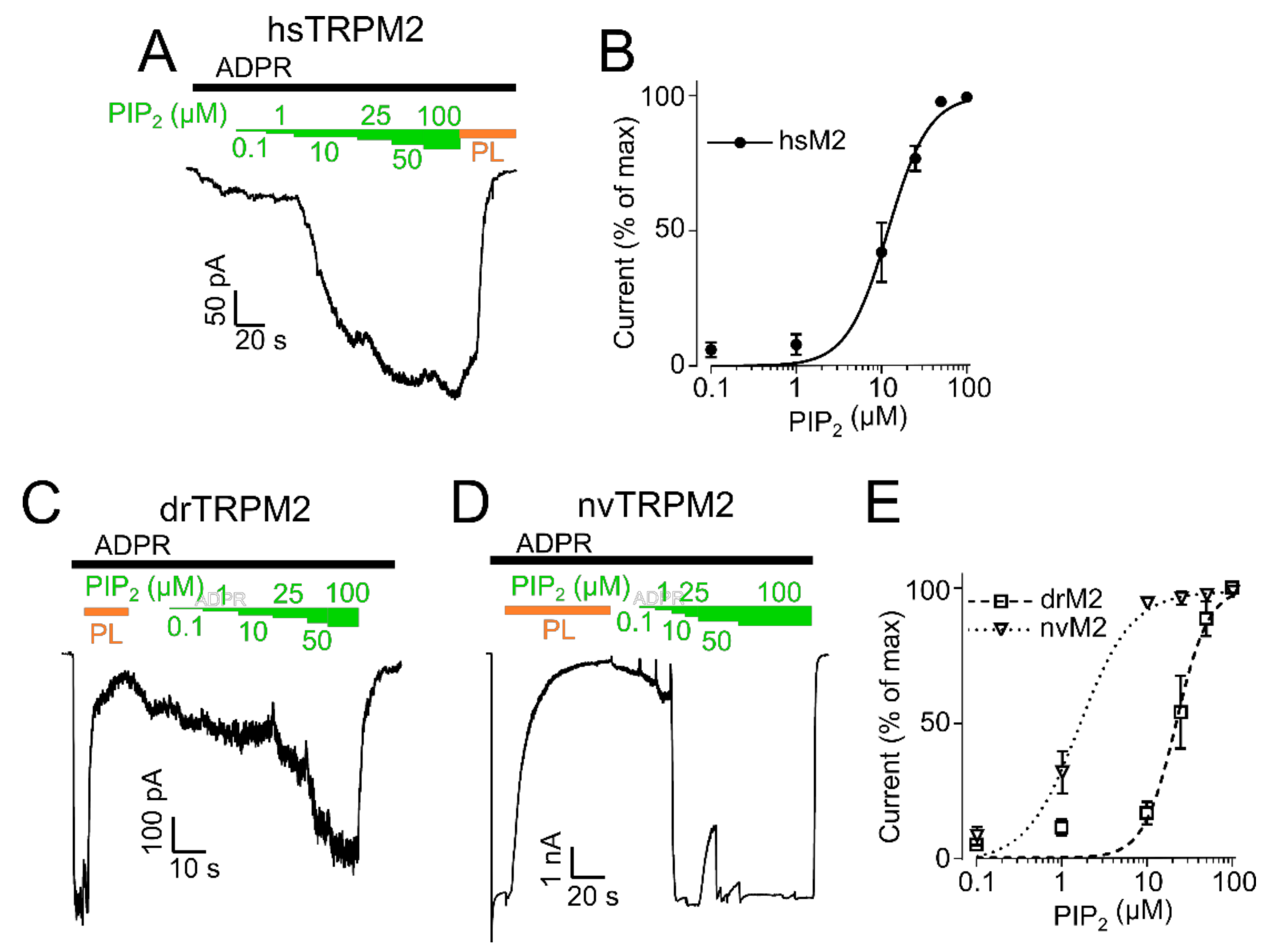
Since the initial experiments indicated species-related differences in the sensitivity of TRPM2 orthologues regulated by PIP2, we decided to induce gradual responses by various PIP2 concentrations to determine EC50 values. After hsTRPM2 stimulated with ADPR evoked a stable baseline current, increasing PIP2 concentrations (from 0.1 to 100 µM) were consecutively added (Figure 3A). An EC50 value of 11.9 ± 1.1 µM (Figure 3B) was determined for hsTRPM2. For drTRPM2 and nvTRPM2, a different protocol had to be applied since ADPR (100 µM) in absence of external PIP2 already elicited maximal currents that were not further enhanced by additional PIP2. Hence, polylysine was used to scavenge PIP2 from ADPR-stimulated patches prior to the PIP2 dose-response. Then, PIP2 was gradually replenished through the perfusion system (Figure 3C for drTRPM2 and Figure 3D for nvTRPM2). The EC50 values calculated in this protocol were 22.3 ± 2.0 µM for drTRPM2 and 2.1 ± 0.2 µM for nvTRPM2 (Figure 3E), however, slow time-dependent current recovery via endogenous free PIP2 may contaminate the effects marginally. These data indicate higher PIP2 sensitivity for nvTRPM2 than for drTRPM2. It should be noted that a direct quantitative comparison of drTRPM2 and nvTRPM2 with hsTRPM2 was not possible because the EC50 values were determined under different conditions. However, hsTRPM2 seems to be the least sensitive towards PIP2 which was also evident as washout of externally applied PIP2 caused a reduction in hsTRPM2 current (Supplementary Figure S1G). The respective EC50 values are the basis for the determination of how various mutations affect the PIP2 sensitivity of each orthologue.
2.3. Residues within the Membrane Interfacial Cavity Are Putative PIP2-Interaction Sites
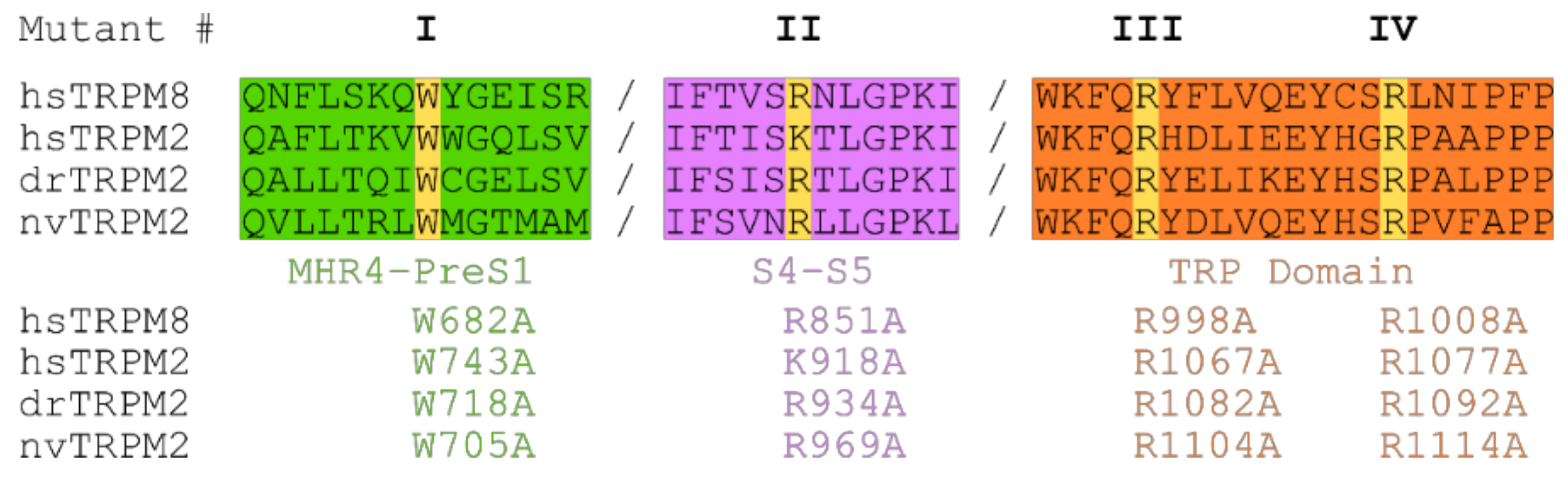
Next, we aimed to identify potential PIP2 interaction sites and their contribution to PIP2-dependent channel regulation. In the cation channel TRPM8, the closest relative of TRPM2, several amino acid residues have been associated with such a role [27,32,33]. Most of them are positively charged and located within the membrane interfacial cavity which in turn is formed by the pre-S1 domain, the junction between S4 and S5, the TRP domain, and MHR4 from the adjacent subunit. In analogy, we chose four residues most critical in TRPM8 and performed a substitution with alanine at corresponding positions of each of the three species variants of TRPM2. Figure 4 specifies the exact position of each of these point mutations which we designate as mutations I to IV for each species for simplicity (e.g., homologous exchange of W743A in hsTRPM2, W718A in drTRPM2 and W705A in nvTRPM2 are all referred to as mutation I).
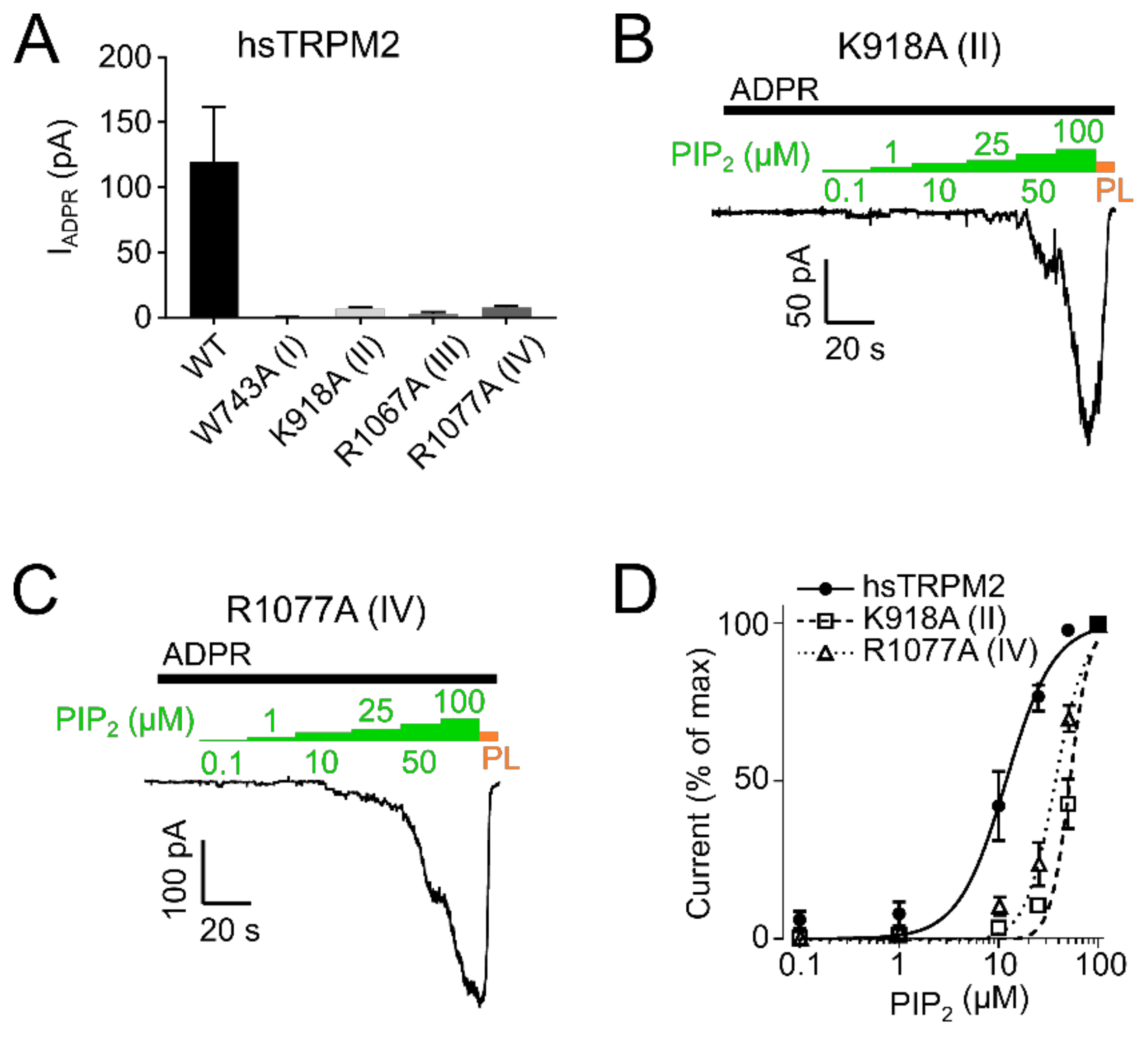
In hsTRPM2 we found that W743A (I) showed a complete absence of ADPR induced currents and was therefore considered non-functional (Figure 5A). Analysis of R1067A (III) showed that ADPR induced either negligible or no response (Figure 5A) and was also unaffected by additional application of up to 100 µM PIP2 (Figure S1B), thereby preventing a quantitative analysis of the PIP2 effects. K918A (II) and R1077A (IV) showed substantial decreases in the current amplitudes (Figure 5A).
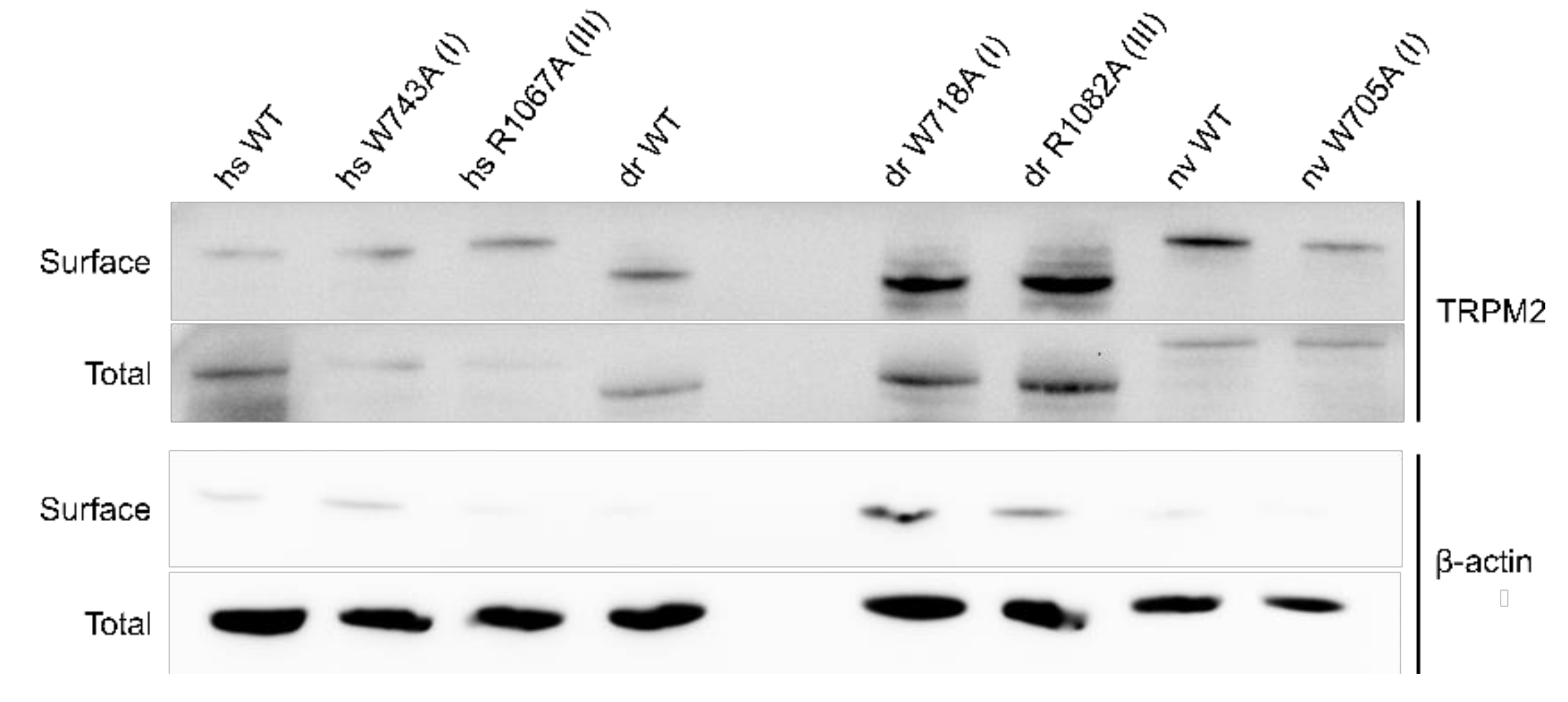
As a control, membrane expression was tested with biotinylation assays showing no significant differences in membrane expression between hsTRPM2 WT and mutants (Figure 6).
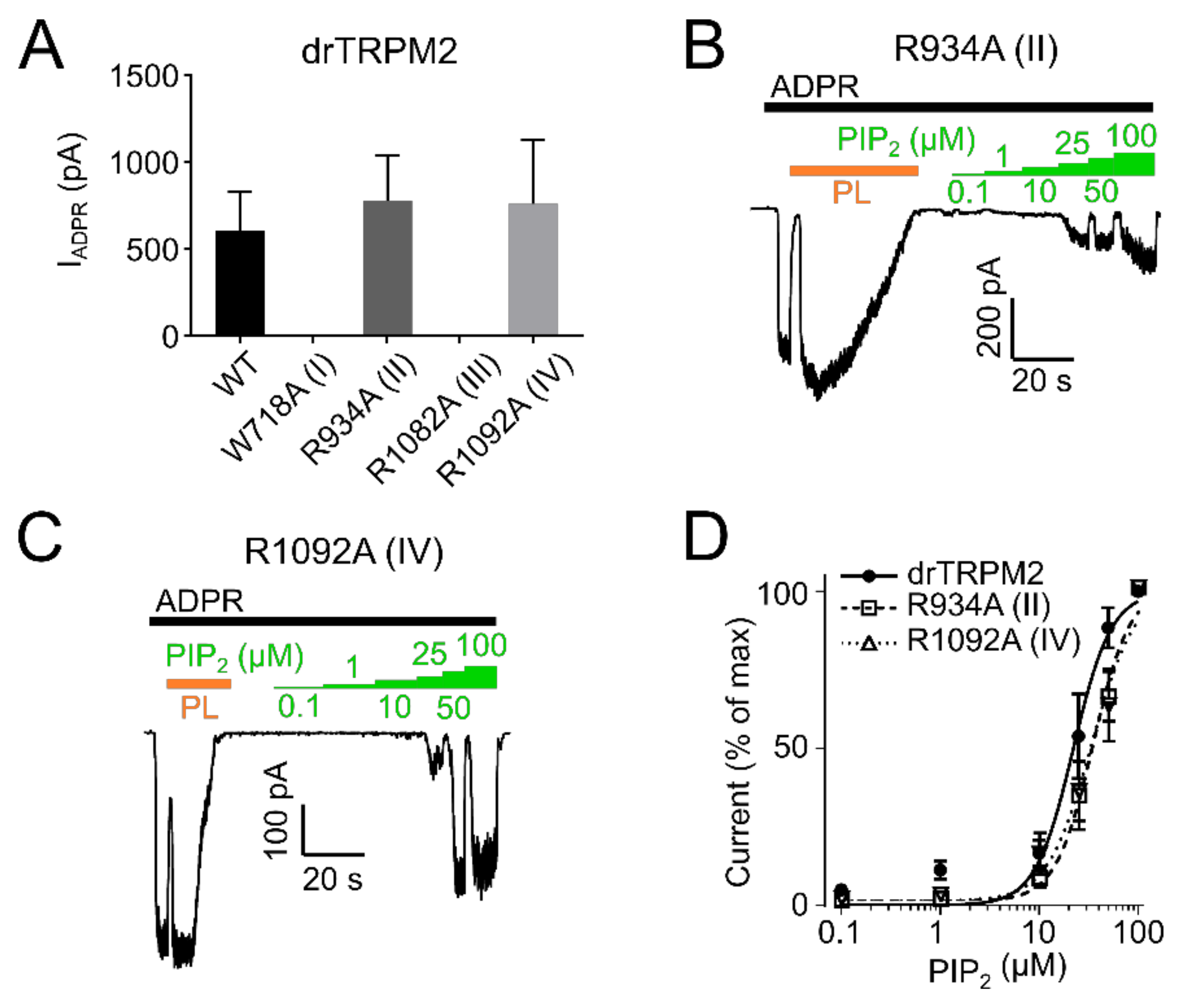
To evaluate whether the decreased current was due to decreased PIP2 sensitivity, we employed dose-response measurements and determined EC50 values for PIP2. PIP2 EC50 for hsTRPM2 K918A (II) and R1077A (IV) were 52.2 µM and 36.3 µM respectively, displaying a pronounced rightward shift when compared with wild type hsTRPM2 (11.9 µM) (Figure 5B–D). Therefore, K918 (II) and R1077 (IV) seem to be crucial for PIP2 interaction, and removal of either residue causes substantially reduced PIP2 sensitivity. Next, we tested the corresponding residues in drTRPM2. W718A (I) and R1082A (III) did not show any currents in response to the application of ADPR (Figure 7A).
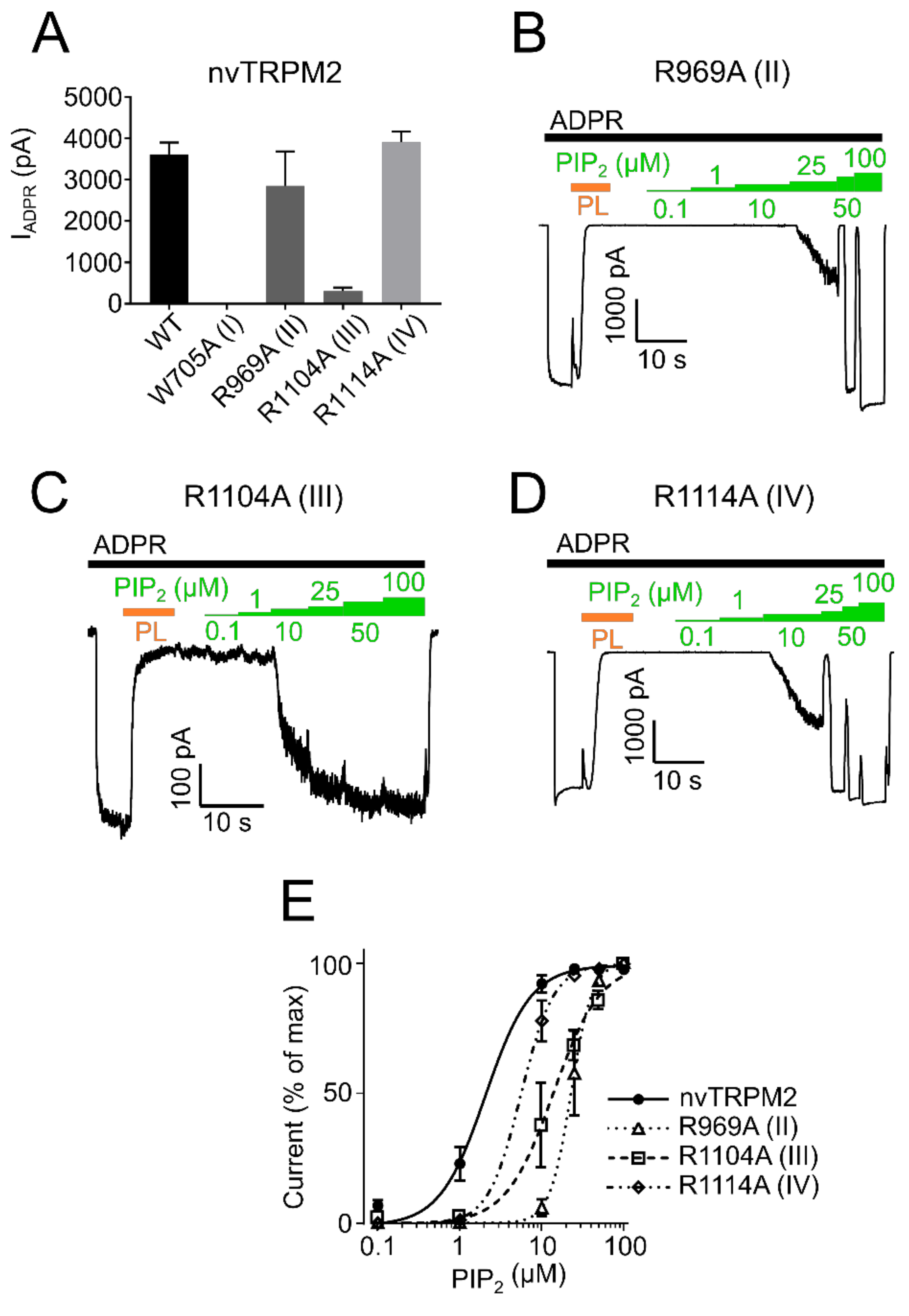
However, since surface expression was increased compared to wild type (Figure 6) the mutants appear to be non-functional and were not further studied. R934A (II) and R1092A (IV) showed robust currents with a slightly higher current amplitude compared with WT drTRPM2 (Figure 7A). Both mutants displayed decreases in sensitivity to PIP2, that trended towards rightward shifts of the EC50 values with 34.6 µM for R934A (II) and 33.6 µM for R1092A (IV) but were not statistically significant when compared with wild type drTRPM2 (22.3 µM) (Figure 7B–D). For nvTRPM2 W705A (I) no currents were observed in the presence of ADPR similar to a mutation I in hsTRPM2 and drTRPM2 (Figure 8A). Even though surface expression for W705A (I) is reduced but still prominent, the mutant was considered non-functional and not subjected to further investigations (Figure 6). R1104A (III) displayed minuscule current amplitudes while R969A (II) and R1114A (IV) showed ADPR induced currents comparable to wild-type nvTRPM2 (Figure 8A). Analysing PIP2 sensitivity of the three functional mutants shows a distinct right shift with R969A (II) displaying the most prominent effect (Figure 8B–E). The EC50 for R969A (II), R1104A (III), and R1114A (IV) was 22.9 µM, 14.2 µM, and 5.6 µM respectively while the wild type was 2.1 µM (Figure 8E). Please note that the PIP2 dose-response curves in all three channel orthologues do not saturate in all mutants and EC50 values might be even larger than stated in this study.
These data suggest that positively charged amino acids within the membrane interfacial cavity are important for PIP2 activation and potentially facilitate PIP2 interaction within the channel. Interestingly, mutation I (conserved tryptophan within the MHR4-PreS1 domain) causes loss of function in all three channel orthologues indicating the great importance of this residue. Even though this could potentially indicate the significance of this residue for PIP2 interaction and therefore channel function, the loss of function could also be explained by protein misfolding or conformational changes. In addition, mutation II within the S4–S5 linker region had the strongest effects on PIP2 sensitivity in all three channel orthologues and could therefore represent a key residue for PIP2 interaction.
3. Discussion
In this study, we investigated the dependence of the TRPM2 function on PIP2. Owing to pronounced species-specific variability in TRPM2 gating we investigated three-channel orthologues of TRPM2 (human, zebrafish, and sea anemone) heterologously expressed in mammalian HEK-293 cells via inside-out patch-clamp recordings. As the main finding, we demonstrated that PIP2 essentially contributes to ADPR-induced activation of TRPM2 in all three species. However, there were remarkable differences in the extent to which currents could be restored after PIP2 depletion. In hsTRPM2, there was no restoration at all unless intracellular Ca2+ was supplemented in high concentrations along with PIP2. Our results indicate that PIP2 interacts with residues within the membrane interfacial cavity of TRPM2 and facilitates channel activation in the presence of ADPR and Ca2+.
3.1. PIP2 Is a Necessary Cofactor for TRPM2 Function
PIP2 dependence of hsTRPM2 and nvTRPM2 expressed in Xenopus oocytes was demonstrated previously showing TRPM2 currents suppressed by polylysine application and fully reactivated via PIP2 [30,31]. In the mammalian expression system employed in the present study, removal of endogenous PIP2 by polylysine followed by exogenous PIP2 application was not sufficient for a re-activation of hsTRPM2 even at high PIP2 concentrations (100 µM). The most likely explanation for the discrepancy in our opinion is the huge differences in free Ca2+. In fact, increasing intracellular free Ca2+ from 1 µM to non-physiological concentrations of 100 µM results in a significant current recovery in hsTRPM2 via PIP2. In contrast, nvTRPM2 was able to fully and rapidly restore currents via exogenously applied PIP2 and physiological calcium concentrations (1 µM), whereas drTRPM2 only recovered partially. This may be explained by the increased calcium sensitivity and conductance of nvTRPM2 compared with hsTRPM2 [23,31]. In this scenario, physiological calcium concentrations in combination with PIP2 are sufficient to reactivate the channel whereas hsTRPM2 requires higher calcium concentrations for reactivation. This seems plausible as it was suggested that PIP2 and calcium may stabilize the open state of nvTRPM2 independently [31] and therefore both calcium and PIP2 may be required in sufficient amounts to support the reactivation of TRPM2 channels. Alternatively, it was proposed that the PIP2 head group itself may be involved in calcium-binding of nvTRPM2 [31] which also highlights the co-dependent action of calcium and PIP2 in activating TRPM2. Moreover, endogenous PIP2 concentrations may be insufficient to fully activate hsTRPM2, especially during stimulation under moderate Ca2+ concentrations, since the application of PIP2 further increases currents in hsTRPM2. Interestingly, nvTRPM2 and drTRPM2 already displayed maximal currents at endogenous PIP2 levels within the membrane patch, which was not further increased by additional PIP2 application. However, it should be noted that using lower ADPR and Ca2+ concentrations when stimulating drTRPM2 and nvTRPM2 could result in reduced starting open probabilities which may result in further stimulation upon PIP2 addition.
Therefore, hsTRPM2 might be more susceptible to regulation by changes in intracellular PIP2 concentrations, while nvTRPM2 might be saturated at most times due to its high PIP2 sensitivity and therefore less susceptible to changes at endogenous PIP2 levels. DrTRPM2 seems to be right in between those two extremes since it is fully activated at endogenous PIP2 levels but is unable to fully recover after PIP2 removal via polylysine.
3.2. Structural Elements of TRPM2 for PIP2 Interaction
Interaction of PIP2 with ion channels is believed to rely on one of two different models or a combination of both [36]. The first model proposes more than 10 basic amino acids in close proximity by sequence generating a strong electrostatic site for nonspecific interactions with negatively charged head groups of phosphoinositides as is the case for the basic effector domain (BED) in MARCKS (myristoylated alanine-rich C kinase substrate). The second model is based on the pleckstrin homology domain (PH domain) in e.g., PLCδ1 (phospholipase-Cδ1) which represents multiple basic amino acids that are not adjacent to one another within the amino acid sequence but are part of a binding pocket within the folded protein. Therefore, BEDs represent unspecific electrostatic binding areas for phosphoinositides while PH domains represent a more specific binding pocket in addition to the electrostatic forces of the basic amino acids [36]. As a result, BEDs bind various phosphoinositides due to their low specificity whereas PH domains are more likely to bind to a specific phosphoinositide for steric reasons. In TRPM2, a cluster of basic amino acids in the primary sequence is absent. Therefore, PIP2 binding of TRPM2 may be facilitated via basic amino acids as part of a binding pocket within the folded protein even though phosphoinositide specificity is not limited to PI(4,5)P2 alone as PI(3,4,5)P3 and in part PI(3,4)P2 (drTRPM2) restore channel activity. Our finding that basic amino acids within the membrane interfacial cavity are necessary for PIP2 interaction is in agreement with this scenario. This is supported by a cryo-electron microscopy study of nvTRPM2 showing a phospholipid density within the membrane interfacial cavity [31]. In addition, this area represents a conserved region, as the binding site of PIP2 in TRPM8 is located in a similar site and PIP2 binding is facilitated by conserved residues [27,32,33]. It should be noted that the corresponding residue for mutant IV in TRPM8 does not directly interact with PIP2 [32] even though mutation of this residue caused the strongest rightward shift of PIP2 EC50 in TRPM8 [33] and may therefore affect activation by PIP2 indirectly. Furthermore, we cannot exclude that other residues examined in this study (e.g., mutant I and III in hsM2 and drM2), may also be involved in PIP2 binding, as the loss of function in these mutants may suggest a crucial role in PIP2 coordination or impairment of the channel protein by other mechanisms (e.g., misfolding). Interestingly, the two corresponding residues in TRPM8 coordinate direct binding between pre-S1 and TRP-like domains mediating gating and functional regulation by PIP2 [27]. Therefore, loss of function in hsTRPM2 and drTRPM2 of mutants I and III may be a result of a missing N-C interaction facilitated via PIP2. Moreover, mutant III could also represent a key residue for PIP2 binding. This is because mutation of this basic residue may disrupt PIP2 binding completely in hsTRPM2 and drTRPM2 due to their lower PIP2 sensitivity resulting in loss of function of the channel, whereas in nvTRPM2 PIP2 binding via the remaining residues may still be possible due to the strong PIP2 sensitivity of nvTRPM2. Finally, we cannot rule out potential further residues involved in PIP2 binding that have not been investigated in this study or that the mutations performed in this study may also affect ADPR and or Ca2+ responses. Future studies investigating the co-structure of TRPM2 in complex with PIP2 will likely provide definitive proof for the PIP2 binding site in TRPM2.
3.3. Physiological Role of PIP2 Interaction with TRPM2
PIP2 is a minor but specific component of the plasma membrane in eukaryotic cells [37]. This allows ion channels that are specifically activated by PIP2 to remain inactive while embedded in intracellular membranes during synthesis and transport to the plasma membrane [37,38]. This feature may also be true for TRPM2 as this channel requires PIP2 for full activation. In addition, PIP2 may act as a modulator of TRPM2 activity in diverse physiological processes. Therefore, TRPM2 activity may be modulated by changes in PIP2 levels within the plasma membrane. This may be achieved by activation of receptors coupled to lipid kinases or PLC resulting in transient changes of specific membrane lipids sufficient to modulate TRPM2 function. This is of particular interest for ion channels with low PIP2 affinity as the membrane concentration of lipids is probably changed by less than one order of magnitude during receptor activation of enzymes [38]. Therefore, hsTRPM2 with its low PIP2 affinity might be more susceptible to such regulatory changes compared with drTRPM2 while nvTRPM2 may not be regulated by changes in PIP2 levels due to its significantly higher affinity. In hsTRPM2, depletion of PIP2 may stop the positive feedback loop of excessive Ca2+ influx. As a result of activation of G protein-coupled receptors (GPCR), PLC cleaves PIP2 into IP3 and diacylglycerol, reducing Ca2+ influx through TRPM2 during increased Ca2+ elevation as a result of IP3-induced Ca2+ release from internal storages [39]. It is also worth mentioning that TRPM2 has been implicated in Alzheimer’s disease [40,41]. Interestingly, PLC is increased and accumulates in the brains of Alzheimer’s disease patients which aligns well with a report of reduced levels of phosphoinositide in these patients [42,43,44]. Therefore, it would be of great interest to test the influence of TRPM2 regulation via phosphoinositides in Alzheimer’s disease.
In conclusion, we have demonstrated that PIP2 enhances TRPM2 activity in all three species by means of homologous, highly conserved structural elements. The gradual requirement for the phospholipid varies remarkably between the channel orthologues, however, the uniform principle strongly contrasts the divergence that has been shown for the mechanisms used by ADPR when it gates TRPM2 in various species. The study contributes to our evolving understanding of complex modulatory processes in TRPM2 channel activation and their structural basis. These may have developed during the evolution of TRPM2 to fulfil divergent functional roles in different species and habitats.
4. Materials and Methods
4.1. Molecular Biology
Subcloning of the TRPM2 cDNA from human, zebrafish (Danio rerio), and sea anemone (N. vectensis) into the modified pIRES-hrGFP-2a vector was described previously [13,23]. Site-directed mutagenesis was performed via the Quikchange mutagenesis kit according to the manufacturer’s instructions (Agilent, Santa Clara, CA, USA), and sequences were verified (MWG-Biotech, Ebersberg, Germany). Custom-made oligonucleotides were acquired from MWG-Biotech. For immunohistological detection in surface expression experiments, wild-type and mutant channels were C-terminally fused with a triple hemagglutinin-tag (3×HA-tag) as previously described [16].
4.2. Biotinylation
Biotinylation assays were performed using the Pierce Cell Surface Protein Insolation Kit according to the manufacturer’s instructions (Thermo Fisher Scientific, Dreieich, Germany). In brief, transfected cells were grown to about 90% confluency and subsequently biotinylated and lysed. Samples (600 µg) were then incubated with NeutrAvidin beads while a small aliquot of total cell lysate was kept as a control. Elution was performed using SDS sample buffer and subjected to SDS-PAGE and Western blot analysis. β-actin was used as a control to rule out biotinylation of cytosolic proteins after cell damage. β-actin was detected via primary mouse-anti-β-actin antibody (1:1000, Cell Signaling, Frankfurt, Germany) and rabbit-anti-mouse-HRP conjugated secondary antibody (1:1000, Agilent). HA-tagged TRPM2 protein was detected via primary monoclonal-mouse-anti-HA antibody (1:1000, Sigma-Aldrich, Darmstadt, Germany) and rabbit-anti-mouse-HRP conjugated secondary antibody (1:1000, Agilent). Detection was visualized using the enhanced chemiluminescence detection system (ECL, Amersham Bioscience, Freiburg, Germany).
4.3. Cell Culture and Transfection
Human embryonic kidney (HEK-293) cells were purchased from the German Collection of Microorganisms and Cell Cultures. Cell culture was carried out in DMEM media (Biochrome, Berlin, Germany) supplemented with 4 mM L-glutamine, 2 mM sodium pyruvate, and 10% (v/v) foetal calf serum (Biochrome). Wild-type or mutant TRPM2 channels were heterologously expressed in HEK-293 cells after transient transfection of the corresponding cDNA using the FuGene 6 transfection reagent (Promega, Walldorf, Germany) according to the manufacturer’s protocol. The transfected cells were incubated for 24 h at 37 °C and 5% CO2. Afterwards, the cells were harvested for biotinylation assay and Western blot analysis. Alternatively, the cells were seeded on cell culture dishes at a suitable dilution and further incubated for 3–4 h. Then, patch-clamp experiments were performed with cells visibly positive for EGFP-expression.
4.4. Patch Clamp
Inside-Out Patch Clamp recordings were performed at room temperature and membrane potential of −60 mV using an Axopatch 200B amplifier and digitised via a Digidata 1440 A. Data were recorded on a personal computer with Clampex 10.7 and analysed using Clampfit 10.7 (Axon Instruments, Molecular Devices, San Jose, CA, USA). The standard bath solution contained (in mM) 145 CsCl, 8 NaCl, 2 MgCl2, 10 HEPES, pH 7.2 (CsOH) and the Ca2+ concentration was adjusted to 1 µM (0.886 mM Ca2+, 1 mM Cs-EGTA) or 100 µM (1.1 mM Ca2+, 1 mM Cs-EGTA). The pipette solution contained (in mM) 140 NaCl, 1.2 MgCl2, 1.2 CaCl2, 5 KCl, 10 HEPES, pH 7.4. For the application of ADPR, phosphoinositides, and polylysine a rapid solution exchange was performed via an RSC-200 (Bio-Logic Science Instruments, Göttingen, Germany). Patch pipettes were made of borosilicate glass (Hilgenberg, Malsfeld, Germany) and had tip resistances between 2 and 4 MΩ. A gap-free acquisition mode was used with analogous filtering at 2 kHz. ADPR (#A0752, Sigma-Aldrich, Darmstadt, Germany) and poly-L-lysine hydrobromide (#P2636, Sigma-Aldrich, Darmstadt, Germany) were diluted into the bath solution from 100 mM and 15 µg/mL aqueous stock respectively. 100 µM ADPR was used for activation of drTRPM2 and nvTRPM2 while 300 µM ADPR was used for hsTRPM2. Phosphoinositides (PI(4)P #10007711, PI(3,4)P2 #10008400, PI(4,5)P2 #64910, PI(3,4,5)P3 #10007764; Cayman Chemical, Tallinn, Estonia) were diluted into the bath solution from a 2.5 mM aqueous stock.
4.5. Data Analysis
Data are expressed as mean ± standard error of the mean (SEM). The numbers of experiments are presented as n. Graphs were generated via GraphPad Prism 6 and Inkscape 0.92. PIP2 dose-response recordings were calculated as a percentage of the maximum current obtained at 100 µM PIP2. Statistical comparisons were made using one-way ANOVA with Dunn’s multiple comparison test and paired Student’s t-test and differences were considered significant at * p < 0.05, ** p < 0.01, ns = not significant.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/ijms22094637/s1, Figure S1: PIP2 and calcium effects on hsTRPM2 and drTRPM2 activity. Figure S2: ADPR dose-response and current rundown of hs-, dr- and nvTRPM2.
Author Contributions
The study was conceptualised by all three authors. D.B. performed the research, obtained and analysed the data. D.B. drafted and finalised the manuscript. F.J.P.K. and A.L. reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by the Deutsche Forschungsgemeinschaft (DFG, Grant KU 2271/4-2 to F.J.P.K.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on reasonable request from the corresponding author.
Acknowledgments
Authors acknowledge the expert technical assistance of Marina Wolf and Manfred Dewor.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kuhn, F.J.P. Structure-function relationship of TRPM2: Recent advances, contradictions, and open questions. Int. J. Mol. Sci. 2020, 21, 6481. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Inamura, K.; Miyake, A.; Mochizuki, S.; Yokoi, H.; Matsushime, H.; Furuichi, K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science 2001, 293, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Csanady, L.; Torocsik, B. Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but intracellularly of the gate. J. Gen. Physiol. 2009, 133, 189–203. [Google Scholar] [CrossRef]
- McHugh, D.; Flemming, R.; Xu, S.Z.; Perraud, A.L.; Beech, D.J. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J. Biol. Chem. 2003, 278, 11002–11006. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef]
- Sun, L.; Yau, H.Y.; Wong, W.Y.; Li, R.A.; Huang, Y.; Yao, X. Role of TRPM2 in H(2)O(2)-induced cell apoptosis in endothelial cells. PLoS ONE 2012, 7, e43186. [Google Scholar] [CrossRef]
- Inamura, K.; Sano, Y.; Mochizuki, S.; Yokoi, H.; Miyake, A.; Nozawa, K.; Kitada, C.; Matsushime, H.; Furuichi, K. Response to ADP-ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J. Membr. Biol. 2003, 191, 201–207. [Google Scholar] [CrossRef]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef]
- Tan, C.H.; McNaughton, P.A. TRPM2 and warmth sensation. Pflug. Arch. 2018, 470, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Toth, B.; Iordanov, I.; Csanady, L. Selective profiling of N- and C-terminal nucleotide-binding sites in a TRPM2 channel. J. Gen. Physiol. 2020, 152. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, F.J.P.; Watt, J.M.; Potter, B.V.L.; Luckhoff, A. Different substrate specificities of the two ADPR binding sites in TRPM2 channels of Nematostella vectensis and the role of IDPR. Sci. Rep. 2019, 9, 4985. [Google Scholar] [CrossRef]
- Kuhn, F.J.P.; Ehrlich, W.; Barth, D.; Kuhn, C.; Luckhoff, A. Functional importance of NUDT9H domain and N-terminal ADPR-binding pocket in two species variants of vertebrate TRPM2 channels. Sci. Rep. 2019, 9, 19224. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Roth, B.; Lu, W.; Du, J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. Elife 2019, 8. [Google Scholar] [CrossRef]
- Huang, Y.; Winkler, P.A.; Sun, W.; Lü, W.; Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 2018. [Google Scholar] [CrossRef]
- Kuhn, F.J.; Kuhn, C.; Winking, M.; Hoffmann, D.C.; Luckhoff, A. ADP-Ribose Activates the TRPM2 Channel from the Sea Anemone Nematostella vectensis independently of the NUDT9H domain. PLoS ONE 2016, 11, e0158060. [Google Scholar] [CrossRef]
- Iordanov, I.; Toth, B.; Szollosi, A.; Csanady, L. Enzyme activity and selectivity filter stability of ancient TRPM2 channels were simultaneously lost in early vertebrates. Elife 2019, 8. [Google Scholar] [CrossRef]
- Toth, B.; Iordanov, I.; Csanady, L. Putative chanzyme activity of TRPM2 cation channel is unrelated to pore gating. Proc. Natl. Acad. Sci. USA 2014, 111, 16949–16954. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, F.J.; Luckhoff, A. Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J. Biol. Chem. 2004, 279, 46431–46437. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.L.; Schmitz, C.; Scharenberg, A.M. TRPM2 Ca2+ permeable cation channels: From gene to biological function. Cell Calcium 2003, 33, 519–531. [Google Scholar] [CrossRef]
- Wang, L.; Fu, T.M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, F.J.; Kuhn, C.; Luckhoff, A. Functional characterisation of a TRPM2 orthologue from the sea anemone Nematostella vectensis in human cells. Sci. Rep. 2015, 5, 8032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, A.L.; Song, W.; Sansom, M.S.P. Lipid-dependent regulation of ion channels and G protein-coupled receptors: Insights from structures and simulations. Ann. Rev. Pharmacol. Toxicol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Choquette, D.; Hakim, G.; Filoteo, A.G.; Plishker, G.A.; Bostwick, J.R.; Penniston, J.T. Regulation of plasma membrane Ca2+ ATPases by lipids of the phosphatidylinositol cycle. Biochem. Biophys. Res. Commun. 1984, 125, 908–915. [Google Scholar] [CrossRef]
- Rohacs, T. Phosphoinositide regulation of TRP channels. Handb. Exp. Pharmacol. 2014, 223, 1143–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Cai, R.; Hofmann, L.; Nesin, V.; Hu, Q.; Long, W.; Fatehi, M.; Liu, X.; Hussein, S.; Kong, T.; et al. Direct binding between Pre-S1 and TRP-like domains in TRPP channels mediates gating and functional regulation by PIP2. Cell. Rep. 2018, 22, 1560–1573. [Google Scholar] [CrossRef] [Green Version]
- Otsuguro, K.; Tang, J.; Tang, Y.; Xiao, R.; Freichel, M.; Tsvilovskyy, V.; Ito, S.; Flockerzi, V.; Zhu, M.X.; Zholos, A.V. Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2008, 283, 10026–10036. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Li, W.P.; Rundle, D.; Kong, J.; Akbarali, H.I.; Tsiokas, L. PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol. Cell. Biol. 2005, 25, 8285–8298. [Google Scholar] [CrossRef] [Green Version]
- Toth, B.; Csanady, L. Pore collapse underlies irreversible inactivation of TRPM2 cation channel currents. Proc. Natl. Acad Sci. USA 2012, 109, 13440–13445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Toth, B.; Szollosi, A.; Chen, J.; Csanady, L. Structure of a TRPM2 channel in complex with Ca(2+) explains unique gating regulation. Elife 2018, 7. [Google Scholar] [CrossRef]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363. [Google Scholar] [CrossRef]
- Rohacs, T.; Lopes, C.M.; Michailidis, I.; Logothetis, D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 2005, 8, 626–634. [Google Scholar] [CrossRef]
- Zhang, H.; Craciun, L.C.; Mirshahi, T.; Rohacs, T.; Lopes, C.M.; Jin, T.; Logothetis, D.E. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 2003, 37, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.M.; Zhang, H.; Rohacs, T.; Jin, T.; Yang, J.; Logothetis, D.E. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 2002, 34, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Suh, B.C.; Hille, B. PIP2 is a necessary cofactor for ion channel function: How and why? Annu. Rev. Biophys. 2008, 37, 175–195. [Google Scholar] [CrossRef] [Green Version]
- Hilgemann, D.W.; Feng, S.; Nasuhoglu, C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE 2001, 2001, re19. [Google Scholar] [CrossRef]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and diacylglycerol as second messengers. Biochem. J. 1984, 220, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.F.; Lavine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The transient receptor potential melastatin 2 (TRPM2) channel contributes to beta-amyloid oligomer-related neurotoxicity and memory impairment. J. Neurosci. 2015, 35, 15157–15169. [Google Scholar] [CrossRef] [Green Version]
- Fonfria, E.; Marshall, I.C.; Boyfield, I.; Skaper, S.D.; Hughes, J.P.; Owen, D.E.; Zhang, W.; Miller, B.A.; Benham, C.D.; McNulty, S. Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 2005, 95, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Shimohama, S.; Sasaki, Y.; Fujimoto, S.; Kamiya, S.; Taniguchi, T.; Takenawa, T.; Kimura, J. Phospholipase C isozymes in the human brain and their changes in Alzheimer's disease. Neuroscience 1998, 82, 999–1007. [Google Scholar] [CrossRef]
- Shimohama, S.; Homma, Y.; Suenaga, T.; Fujimoto, S.; Taniguchi, T.; Araki, W.; Yamaoka, Y.; Takenawa, T.; Kimura, J. Aberrant accumulation of phospholipase C-delta in Alzheimer brains. Am. J. Pathol. 1991, 139, 737–742. [Google Scholar] [PubMed]
- Stokes, C.E.; Hawthorne, J.N. Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. J. Neurochem. 1987, 48, 1018–1021. [Google Scholar] [CrossRef]
Figure 1.
PIP2 is a necessary cofactor for TRPM2 activation. Inside-out patch clamp recordings of HEK-293 cells expressing TRPM2 at −60 mV and 1 µM Ca2+ unless stated otherwise. Currents were activated by cytosolic exposure to ADPR (black bar). (A) Representative current trace of hsTRPM2 activated by cytosolic exposure to 300 µM ADPR (black bar) and 25 µM PIP2 (green bar) and blocked by 15 µg/mL polylysine (PL, orange bar). (B) Summary of the effects of ADPR, PIP2, and polylysine on hsTRPM2 currents. Statistical analysis reveals a significant current increase via PIP2 after the initial ADPR induced current plateaued. The current was abolished upon exposure to polylysine and unable to recover via a second PIP2 application. Control (Ctrl) represents standard bath solution in absence of ADPR. (C,D) Representative current trace and statistics of the PIP2 effect following elevated intracellular Ca2+ concentrations (100 µM) for hsTRPM2. Application of 100 µM Ca2+ causes a strong current increase in hsTRPM2 which is only slightly further increased upon external PIP2 addition. Current traces of drTRPM2 (E) and nvTRPM2 (G) exposed to 100 µM ADPR, 25 µM PIP2 and 15 µg/mL polylysine. Statistical analysis shows no significant difference for drTRPM2 (F) and nvTRPM2 (H) between the current observed during exposure to ADPR and the additional application of PIP2. (F) Fractional reactivation of drTRPM2 currents by second PIP2 application after the current was abolished via polylysine while nvTRPM2 (H) fully recovered. Data are presented as mean ± SEM analysed via one-way ANOVA with Dunn’s multiple comparison test; n = 3–8; * p < 0.05, ** p < 0.01, ns = not significant.
Figure 1.
PIP2 is a necessary cofactor for TRPM2 activation. Inside-out patch clamp recordings of HEK-293 cells expressing TRPM2 at −60 mV and 1 µM Ca2+ unless stated otherwise. Currents were activated by cytosolic exposure to ADPR (black bar). (A) Representative current trace of hsTRPM2 activated by cytosolic exposure to 300 µM ADPR (black bar) and 25 µM PIP2 (green bar) and blocked by 15 µg/mL polylysine (PL, orange bar). (B) Summary of the effects of ADPR, PIP2, and polylysine on hsTRPM2 currents. Statistical analysis reveals a significant current increase via PIP2 after the initial ADPR induced current plateaued. The current was abolished upon exposure to polylysine and unable to recover via a second PIP2 application. Control (Ctrl) represents standard bath solution in absence of ADPR. (C,D) Representative current trace and statistics of the PIP2 effect following elevated intracellular Ca2+ concentrations (100 µM) for hsTRPM2. Application of 100 µM Ca2+ causes a strong current increase in hsTRPM2 which is only slightly further increased upon external PIP2 addition. Current traces of drTRPM2 (E) and nvTRPM2 (G) exposed to 100 µM ADPR, 25 µM PIP2 and 15 µg/mL polylysine. Statistical analysis shows no significant difference for drTRPM2 (F) and nvTRPM2 (H) between the current observed during exposure to ADPR and the additional application of PIP2. (F) Fractional reactivation of drTRPM2 currents by second PIP2 application after the current was abolished via polylysine while nvTRPM2 (H) fully recovered. Data are presented as mean ± SEM analysed via one-way ANOVA with Dunn’s multiple comparison test; n = 3–8; * p < 0.05, ** p < 0.01, ns = not significant.

Figure 2.
Influence of high calcium and PIP2 derivatives on TRPM2 current restoration. Recordings of HEK-293 cells expressing TRPM2 at −60 mV in the inside-out patch clamp configuration. Currents were activated by cytosolic exposure to ADPR (black bar) and PIP2 (25 µM) (green bar). Representative current trace and statistics of hsTRPM2 (A,B) and drTRPM2 (C,D) at high Ca2+ concentrations (100 µM). High Ca2+ in presence of PIP2 and ADPR (blue bar) causes current restoration after the current was inhibited via 15 µg/mL polylysine (PL, orange bar). Representative recordings and summary of current statistics of the effect of PI derivatives on hsTRPM2 (E,F) and drTRPM2 (G,H) currents using 1 µM cytosolic Ca2+. Statistical analysis was performed via paired Student’s t-test and data presented as mean ± SEM; n = 4–7; * p < 0.05.
Figure 2.
Influence of high calcium and PIP2 derivatives on TRPM2 current restoration. Recordings of HEK-293 cells expressing TRPM2 at −60 mV in the inside-out patch clamp configuration. Currents were activated by cytosolic exposure to ADPR (black bar) and PIP2 (25 µM) (green bar). Representative current trace and statistics of hsTRPM2 (A,B) and drTRPM2 (C,D) at high Ca2+ concentrations (100 µM). High Ca2+ in presence of PIP2 and ADPR (blue bar) causes current restoration after the current was inhibited via 15 µg/mL polylysine (PL, orange bar). Representative recordings and summary of current statistics of the effect of PI derivatives on hsTRPM2 (E,F) and drTRPM2 (G,H) currents using 1 µM cytosolic Ca2+. Statistical analysis was performed via paired Student’s t-test and data presented as mean ± SEM; n = 4–7; * p < 0.05.

Figure 3.
PIP2 sensitivity of TRPM2. PIP2 dose-response recordings of inside-out patches of HEK-293 cells expressing hsTRPM2 (A), drTRPM2 (C), and nvTRPM2 (D). ADPR (black bar) was used to initiate TRPM2 currents followed by increasing concentrations of PIP2 (green bars). Polylysine (PL, orange bar) was utilized to scavenge PIP2. Summary of the PIP2 dose-response recordings calculated as a percentage of the maximum current obtained at 100 µM PIP2 (hsTRPM2 (B), drTRPM2, and nvTRPM2 (E)). Data were analysed from five to six independent experiments.
Figure 3.
PIP2 sensitivity of TRPM2. PIP2 dose-response recordings of inside-out patches of HEK-293 cells expressing hsTRPM2 (A), drTRPM2 (C), and nvTRPM2 (D). ADPR (black bar) was used to initiate TRPM2 currents followed by increasing concentrations of PIP2 (green bars). Polylysine (PL, orange bar) was utilized to scavenge PIP2. Summary of the PIP2 dose-response recordings calculated as a percentage of the maximum current obtained at 100 µM PIP2 (hsTRPM2 (B), drTRPM2, and nvTRPM2 (E)). Data were analysed from five to six independent experiments.

Figure 4.
Sequence alignment of TRPM2 and TRPM8. Sequence alignment of the membrane interfacial cavity (formed by: Pre-S1 domain and MHR4 from the adjacent subunit (green), junction between S4 and S5 (purple) and TRP domain (orange)) of hs-, dr- and nvTRPM2 and hsTRPM8. Target positions (yellow) within MHR4-PreS1 labelled mutant I (hsW743, drW718, and nvW705), S4–S5 linker labelled mutant II (hsK918, drR934, and nvR969) and TRP domain separated in mutant III (hsR1067, drR1082, nvR1104) and mutant IV (hsR1077, drR1092, nvR1114).
Figure 4.
Sequence alignment of TRPM2 and TRPM8. Sequence alignment of the membrane interfacial cavity (formed by: Pre-S1 domain and MHR4 from the adjacent subunit (green), junction between S4 and S5 (purple) and TRP domain (orange)) of hs-, dr- and nvTRPM2 and hsTRPM8. Target positions (yellow) within MHR4-PreS1 labelled mutant I (hsW743, drW718, and nvW705), S4–S5 linker labelled mutant II (hsK918, drR934, and nvR969) and TRP domain separated in mutant III (hsR1067, drR1082, nvR1104) and mutant IV (hsR1077, drR1092, nvR1114).

Figure 5.
PIP2 sensitivity of hsTRPM2 membrane interfacial cavity mutant channels. (A) Current amplitudes of wild-type and mutant hsTRPM2 channels in response to 300 µM ADPR. Representative current trace of K918A (II) (B) and R1077A (IV) (C) exposed to increasing PIP2 (green bar) concentrations in the presence of 300 µM ADPR (black bar). 15 µg/mL polylysine (PL, orange bar) was used to block PIP2 mediated currents. (D) Data summary of PIP2 dose-response experiments showing a clear right shift of mutants K918A (II) and R1077A (IV) compared with the wild-type. Data presented as mean ± SEM; n = 5.
Figure 5.
PIP2 sensitivity of hsTRPM2 membrane interfacial cavity mutant channels. (A) Current amplitudes of wild-type and mutant hsTRPM2 channels in response to 300 µM ADPR. Representative current trace of K918A (II) (B) and R1077A (IV) (C) exposed to increasing PIP2 (green bar) concentrations in the presence of 300 µM ADPR (black bar). 15 µg/mL polylysine (PL, orange bar) was used to block PIP2 mediated currents. (D) Data summary of PIP2 dose-response experiments showing a clear right shift of mutants K918A (II) and R1077A (IV) compared with the wild-type. Data presented as mean ± SEM; n = 5.

Figure 6.
Biotinylation assay of TRPM2 expressed in HEK-293 cells. Representative surface and total expression of hs-, dr- and nvTRPM2 WT and point mutations via biotinylation. β-actin was used as the loading control.
Figure 6.
Biotinylation assay of TRPM2 expressed in HEK-293 cells. Representative surface and total expression of hs-, dr- and nvTRPM2 WT and point mutations via biotinylation. β-actin was used as the loading control.

Figure 7.
PIP2 sensitivity of drTRPM2 membrane interfacial cavity mutant channels. (A) ADPR (100 µM, black bar) induced currents of wild-type and mutant drTRPM2 channels. Current trace of R934A (II) (B) and R1092A (IV) (C) perfused with increasing concentrations of PIP2 (green bars). Polylysine (15 µg/mL, PL, orange bar) was used to scavenge PIP2 since even in the absence of external PIP2 the channels are already saturated with natural membrane PIP2. (D) Summary of the presented data showing a right shift in PIP2 sensitivity of mutants R934A (II) and R1092A (IV). Data represented as mean ± SEM; n = 5–6.
Figure 7.
PIP2 sensitivity of drTRPM2 membrane interfacial cavity mutant channels. (A) ADPR (100 µM, black bar) induced currents of wild-type and mutant drTRPM2 channels. Current trace of R934A (II) (B) and R1092A (IV) (C) perfused with increasing concentrations of PIP2 (green bars). Polylysine (15 µg/mL, PL, orange bar) was used to scavenge PIP2 since even in the absence of external PIP2 the channels are already saturated with natural membrane PIP2. (D) Summary of the presented data showing a right shift in PIP2 sensitivity of mutants R934A (II) and R1092A (IV). Data represented as mean ± SEM; n = 5–6.

Figure 8.
PIP2 sensitivity of nvTRPM2 membrane interfacial cavity mutant channels. (A) Current amplitudes induced by ADPR (100 µM, black bar) of wild-type and mutant nvTRPM2 channels. PIP2 (green bars) dose-response recordings of nvTRPM2 R969A (II) (B), R1104A (III) (C) and R1114A (IV) (D) in presence of 100 µM ADPR (black bar). Since maximum currents were already observed in absence of external PIP2, natural PIP2 was scavenged via polylysine (15 µg/mL, PL, orange bar) and subsequently restored via increasing PIP2 concentrations. (E) Data summary of PIP2 sensitivity of mutants R969A (II), R1104A (III), and R1114A (IV) showing dominant right shift compared with wild-type nvTRPM2. Data represented as mean ± SEM; n = 5–6.
Figure 8.
PIP2 sensitivity of nvTRPM2 membrane interfacial cavity mutant channels. (A) Current amplitudes induced by ADPR (100 µM, black bar) of wild-type and mutant nvTRPM2 channels. PIP2 (green bars) dose-response recordings of nvTRPM2 R969A (II) (B), R1104A (III) (C) and R1114A (IV) (D) in presence of 100 µM ADPR (black bar). Since maximum currents were already observed in absence of external PIP2, natural PIP2 was scavenged via polylysine (15 µg/mL, PL, orange bar) and subsequently restored via increasing PIP2 concentrations. (E) Data summary of PIP2 sensitivity of mutants R969A (II), R1104A (III), and R1114A (IV) showing dominant right shift compared with wild-type nvTRPM2. Data represented as mean ± SEM; n = 5–6.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Barth, D.; Lückhoff, A.; Kühn, F.J.P. Species-Specific Regulation of TRPM2 by PI(4,5)P2 via the Membrane Interfacial Cavity. Int. J. Mol. Sci. 2021, 22, 4637. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094637
AMA Style
Barth D, Lückhoff A, Kühn FJP. Species-Specific Regulation of TRPM2 by PI(4,5)P2 via the Membrane Interfacial Cavity. International Journal of Molecular Sciences. 2021; 22(9):4637. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094637
Chicago/Turabian StyleBarth, Daniel, Andreas Lückhoff, and Frank J. P. Kühn. 2021. "Species-Specific Regulation of TRPM2 by PI(4,5)P2 via the Membrane Interfacial Cavity" International Journal of Molecular Sciences 22, no. 9: 4637. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094637
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.