
First Evidence of Past and Present Interactions between Viruses and the Black Soldier Fly, Hermetia illucens

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets and Samples
2.2. Screening BSF Genome for EVEs
2.3. Exogenous Virus Discovery Using Transcriptomic Data
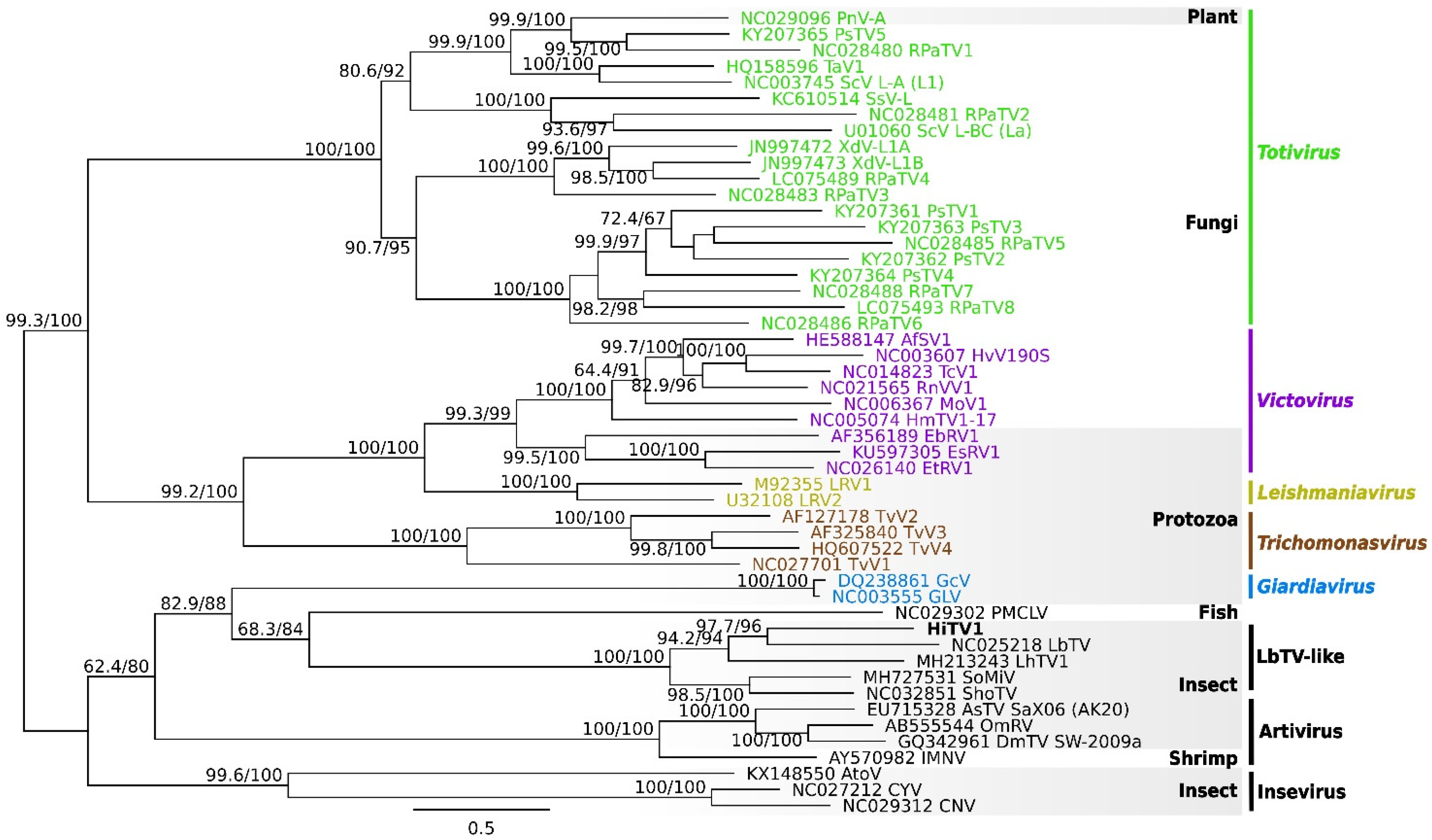
2.4. Phylogeny of Totiviridae
2.5. Molecular Validation of EVE
3. Results and Discussion
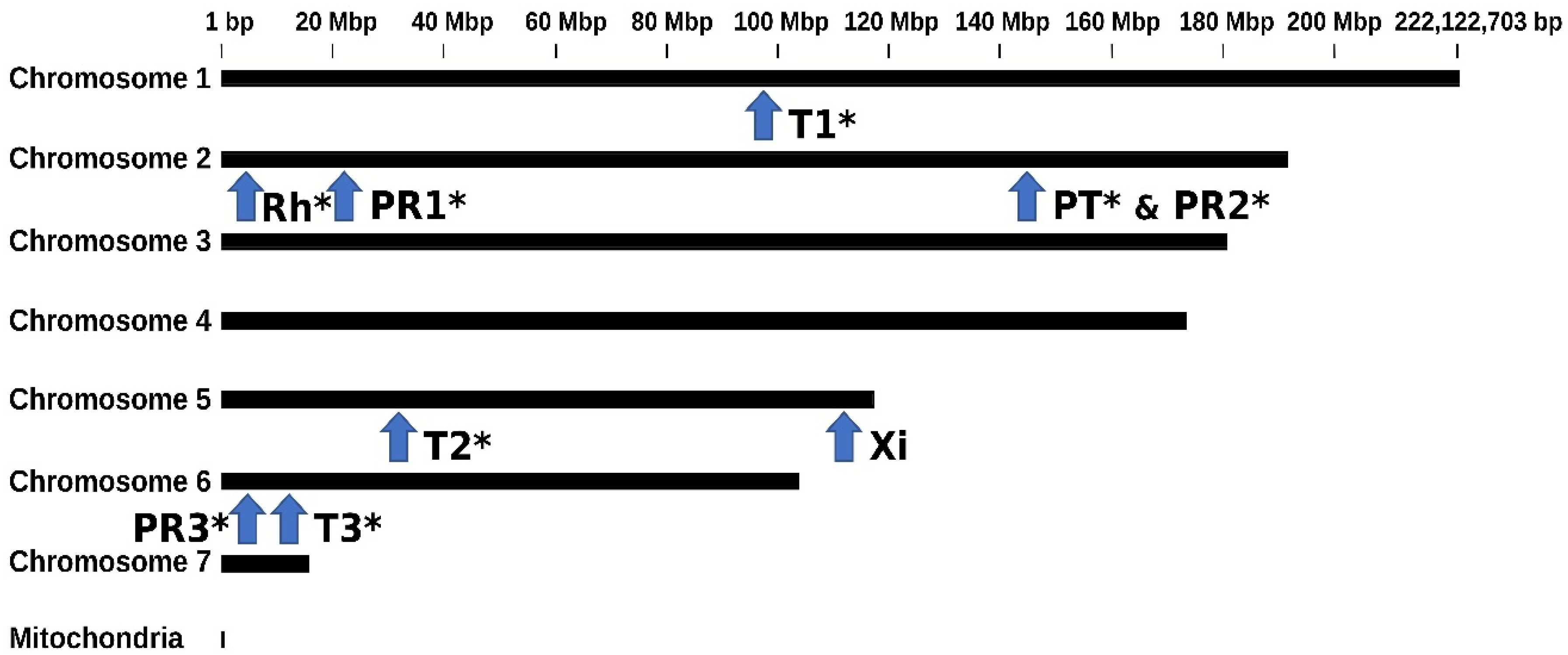
3.1. Orthologous EVE Sequences Found in Three BSF Genomes
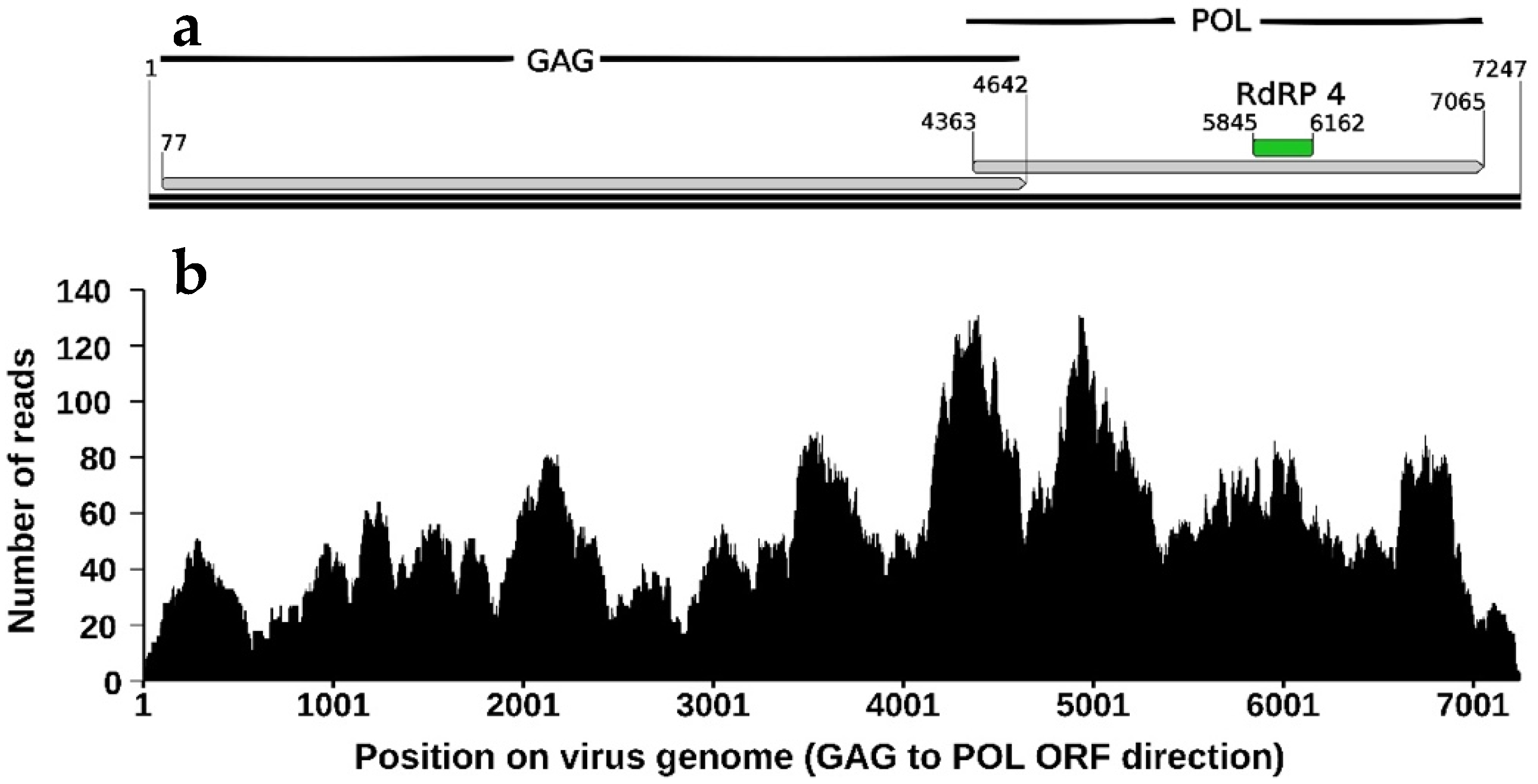
3.2. Description and Phylogeny of HiTV1, an Exogenous Totivirus
3.3. Expression of the Endogenous TotiEVE T1
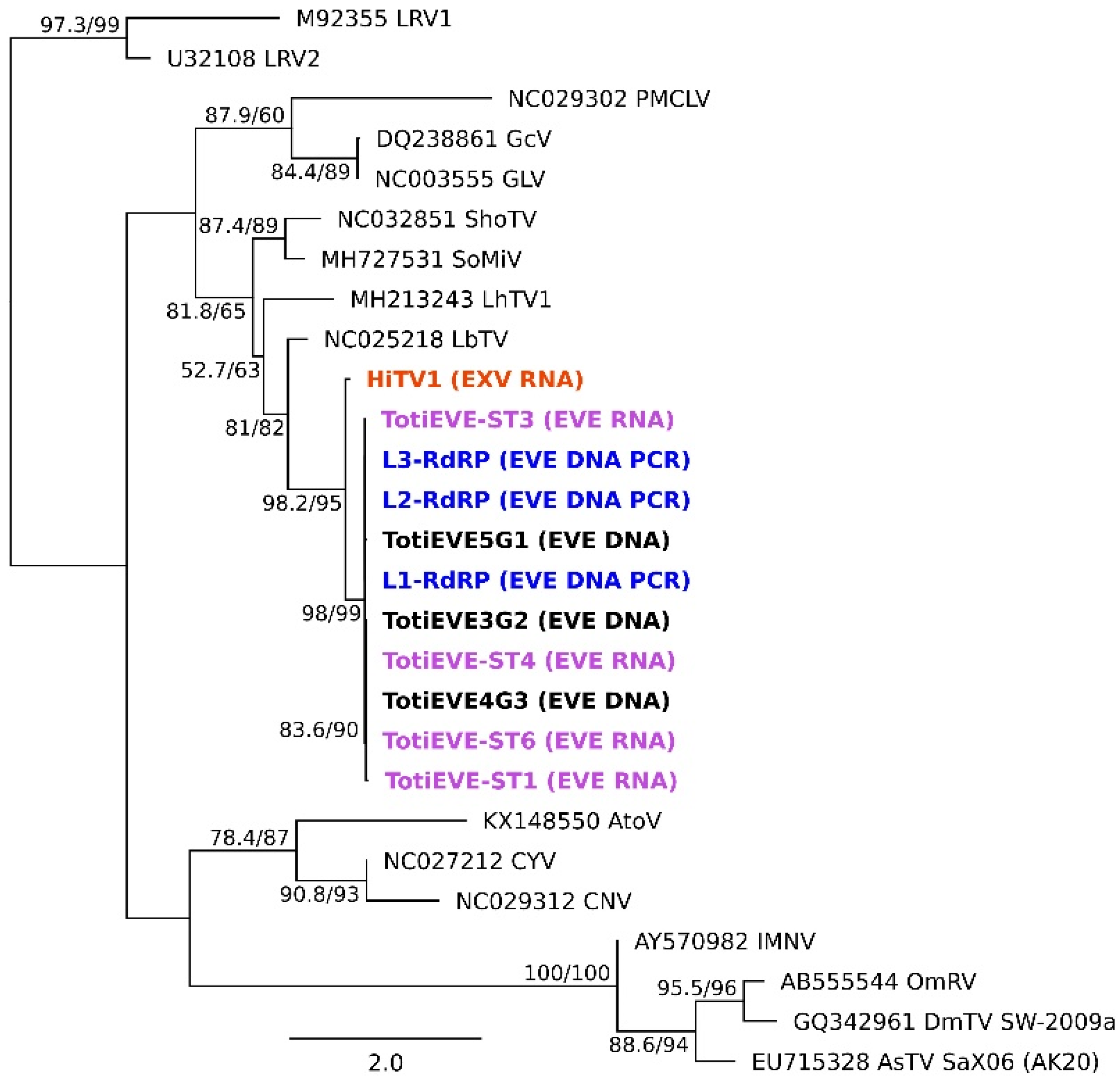
3.4. Phylogenetic Relationships between the Exogenous HiTV1 and TotiEVEs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davidson, E.W. History of insect pathology. In Insect Pathology; Vega, F.E., Kaya, H.K., Eds.; Academic Press: London, UK, 2012; pp. 13–28. ISBN 0123849845. [Google Scholar]
- Maciel-Vergara, G.; Ros, V.I.D. Viruses of insects reared for food and feed. J. Invertebr. Pathol. 2017, 147, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Semberg, E.; de Miranda, J.R.; Low, M.; Jansson, A.; Forsgren, E.; Berggren, Å. Diagnostic protocols for the detection of Acheta domesticus densovirus (AdDV) in cricket frass. J. Virol. Methods 2019, 264, 61–64. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Granberg, F.; Low, M.; Onorati, P.; Semberg, E.; Jansson, A.; Berggren, Å. Virus diversity and loads in crickets reared for feed: Implications for husbandry. Front. Vet. Sci. 2021, 8, 510. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Granberg, F.; Onorati, P.; Jansson, A.; Berggren, Å. Virus prospecting in crickets—discovery and strain divergence of a novel iflavirus in wild and cultivated Acheta domesticus. Viruses 2021, 13, 364. [Google Scholar] [CrossRef] [PubMed]
- Reverberi, M. Edible insects: Cricket farming and processing as an emerging market. J. Insects as Food Feed 2020, 6, 211–220. [Google Scholar] [CrossRef]
- Duffield, K.R.; Hunt, J.; Sadd, B.M.; Sakaluk, S.K.; Oppert, B.; Rosario, K.; Behle, R.W.; Ramirez, J.L. Active and covert infections of cricket iridovirus and acheta domesticus densovirus in reared Gryllodes sigillatus crickets. Front. Microbiol. 2021, 12, 780796. [Google Scholar] [CrossRef]
- Beaurepaire, A.; Piot, N.; Doublet, V.; Antunez, K.; Campbell, E.; Chantawannakul, P.; Chejanovsky, N.; Gajda, A.; Heerman, M.; Panziera, D.; et al. Diversity and global distribution of viruses of the western honey bee, Apis mellifera. Insects 2020, 11, 239. [Google Scholar] [CrossRef]
- Marshall, S.A.; Woodley, N.E.; Hauser, M. The historical spread of the Black Soldier Fly, Hermetia illucens (L.)(Diptera, Stratiomyidae, Hermetiinae), and its establishment in Canada. J. Entomol. Soc. Ontario 2015, 146, 51–54. [Google Scholar]
- Wang, Y.; Shelomi, M. Review of black soldier fly (Hermetia illucens) as animal feed and human food. Foods 2017, 6, 91. [Google Scholar] [CrossRef]
- Van Huis, A. Prospects of insects as food and feed. Org. Agric. 2021, 11, 301–308. [Google Scholar] [CrossRef]
- Tomberlin, J.K.; Huis, A. van Black soldier fly from pest to ‘crown jewel’ of the insects as feed industry: An historical perspective. J. Insects Food Feed. 2020, 6, 1–4. [Google Scholar] [CrossRef]
- Joosten, L.; Lecocq, A.; Jensen, A.B.; Haenen, O.; Schmitt, E.; Eilenberg, J. Review of insect pathogen risks for the black soldier fly (Hermetia illucens) and guidelines for reliable production. Entomol. Exp. Appl. 2020, 168, 432–447. [Google Scholar] [CrossRef]
- Moretta, A.; Salvia, R.; Scieuzo, C.; Di Somma, A.; Vogel, H.; Pucci, P.; Sgambato, A.; Wolff, M.; Falabella, P. A bioinformatic study of antimicrobial peptides identified in the Black Soldier Fly (BSF) Hermetia illucens (Diptera: Stratiomyidae). Sci. Rep. 2020, 10, 16875. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Huang, X.; Tu, F.; Wang, C.; Yang, F. Preparation, antioxidant activity evaluation, and identification of antioxidant peptide from black soldier fly (Hermetia illucens L.) larvae. J. Food Biochem. 2020, 44, e13186. [Google Scholar] [CrossRef]
- Mouithys-Mickalad, A.; Schmitt, E.; Dalim, M.; Franck, T.; Tome, N.M.; van Spankeren, M.; Serteyn, D.; Paul, A. Black soldier fly (Hermetia illucens) larvae protein derivatives: Potential to promote animal health. Animals 2020, 10, 941. [Google Scholar] [CrossRef]
- Xia, J.; Ge, C.; Yao, H. Antimicrobial peptides from black soldier fly (Hermetia illucens) as potential antimicrobial factors representing an alternative to antibiotics in livestock farming. Animals 2021, 11, 1937. [Google Scholar] [CrossRef]
- Tourtois, J.; Ali, J.G.; Grieshop, M.J. Susceptibility of wounded and intact black soldier fly Hermetia illucens (L.) (Diptera: Stratiomyidae) to entomopathogenic nematodes. J. Invertebr. Pathol. 2017, 150, 121–129. [Google Scholar] [CrossRef]
- Lecocq, A.; Joosten, L.; Schmitt, E.; Eilenberg, J.; Jensen, A.B. Hermetia illucens adults are susceptible to infection by the fungus Beauveria bassiana in laboratory experiments. J. Insects Food Feed 2020, 7, 63–68. [Google Scholar] [CrossRef]
- Klüber, P.; Müller, S.; Schmidt, J.; Zorn, H.; Rühl, M. Isolation of bacterial and fungal microbiota associated with Hermetia illucens larvae reveals novel insights into entomopathogenicity. Microorganisms 2022, 10, 319. [Google Scholar] [CrossRef]
- Manu, C.R.; Anitha, N.; Gavas, R.; Poornima Yadav, P.I. Suitability of black soldier fly larvae as host for entomopathogenic nematodes. Indian J. Entomol. 2022, e21179. [Google Scholar] [CrossRef]
- Webster, C.L.; Longdon, B.; Lewis, S.H.; Obbard, D.J. Twenty-five new viruses associated with the Drosophilidae (Diptera). Evol. Bioinform. 2016, 12s2, EBO-S39454. [Google Scholar] [CrossRef] [PubMed]
- Ter Horst, A.M.; Nigg, J.C.; Dekker, F.M.; Falk, B.W. Endogenous viral elements are widespread in arthropod genomes and commonly give rise to PIWI-interacting RNAs. J. Virol. 2019, 93, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pang, R.; Cheng, T.; Xue, L.; Zeng, H.; Lei, T.; Chen, M.; Wu, S.; Ding, Y.; Zhang, J.; et al. Abundant and diverse RNA viruses in insects revealed by RNA-seq analysis: Ecological and evolutionary implications. mSystems 2020, 5, e0039-20. [Google Scholar] [CrossRef]
- Wallace, M.A.; Coffman, K.A.; Gilbert, C.; Ravindran, S.; Albery, G.F.; Abbott, J.; Argyridou, E.; Bellosta, P.; Betancourt, A.J.; Colinet, H.; et al. The discovery, distribution, and diversity of DNA viruses associated with Drosophila melanogaster in Europe. Virus Evol. 2021, 7, veab031. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Belliardo, C. The diversity of endogenous viral elements in insects. Curr. Opin. Insect Sci. 2022, 49, 48–55. [Google Scholar] [CrossRef]
- Patel, M.R.; Emerman, M.; Malik, H.S. Paleovirology—Ghosts and gifts of viruses past. Curr. Opin. Virol. 2011, 1, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Aswad, A.; Katzourakis, A. Paleovirology and virally derived immunity. Trends Ecol. Evol. 2012, 27, 627–636. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Katzourakis, A. Endogenous viruses: Connecting recent and ancient viral evolution. Virology 2015, 479, 26–37. [Google Scholar] [CrossRef]
- Barreat, J.G.N.; Katzourakis, A. Paleovirology of the DNA viruses of eukaryotes. Trends Microbiol. 2022, 30, 281–292. [Google Scholar] [CrossRef]
- Gauthier, J.; Boulain, H.; van Vugt, J.J.F.A.; Baudry, L.; Persyn, E.; Aury, J.-M.; Noel, B.; Bretaudeau, A.; Legeai, F.; Warris, S.; et al. Chromosomal scale assembly of parasitic wasp genome reveals symbiotic virus colonization. Commun. Biol. 2021, 4, 104. [Google Scholar] [CrossRef]
- Cerqueira de Araujo, A.; Huguet, E.; Herniou, E.A.; Drezen, J.-M.; Josse, T. Transposable element repression using piRNAs, and its relevance to endogenous viral elements (EVEs) and immunity in insects. Curr. Opin. Insect Sci. 2022, 50, 100876. [Google Scholar] [CrossRef] [PubMed]
- Vogel, H.; Müller, A.; Heckel, D.G.; Gutzeit, H.; Vilcinskas, A. Nutritional immunology: Diversification and diet-dependent expression of antimicrobial peptides in the black soldier fly Hermetia illucens. Dev. Comp. Immunol. 2018, 78, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Bruno, D.; Brilli, M.; Gianfranceschi, N.; Tian, L.; Tettamanti, G.; Caccia, S.; Casartelli, M. Black soldier fly larvae adapt to different food substrates through morphological and functional responses of the midgut. Int. J. Mol. Sci. 2020, 21, 4955. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Rehman, K.U.; Yu, Y.; Liu, X.; Wang, H.; Tomberlin, J.K.; Sze, S.H.; Cai, M.; Zhang, J.; Yu, Z.; et al. De novo transcriptome sequencing and analysis revealed the molecular basis of rapid fat accumulation by black soldier fly (Hermetia illucens, L.) for development of insectival biodiesel. Biotechnol. Biofuels 2019, 12, 194. [Google Scholar] [CrossRef]
- Zhan, S.; Fang, G.; Cai, M.; Kou, Z.; Xu, J.; Cao, Y.; Bai, L.; Zhang, Y.; Jiang, Y.; Luo, X.; et al. Genomic landscape and genetic manipulation of the black soldier fly Hermetia illucens, a natural waste recycler. Cell Res. 2020, 30, 50–60. [Google Scholar] [CrossRef]
- Xu, Q.; Wu, Z.; Zeng, X.; An, X. Identification and expression profiling of chemosensory genes in Hermetia illucens via a transcriptomic analysis. Front. Physiol. 2020, 11, 720. [Google Scholar] [CrossRef]
- Sherry, S.; Xiao, C.; Durbrow, K.; Kimelman, M.; Rodarmer, K.; Shumway, M.; Yaschenko, E. Ncbi sra toolkit technology for next generation sequence data. In Proceedings of the Plant and Animal Genome XX Conference, San Diego, CA, USA, 14–18 January 2012. [Google Scholar]
- Vicoso, B.; Bachtrog, D. Numerous transitions of sex chromosomes in Diptera. PLoS Biol. 2015, 13, e1002078. [Google Scholar] [CrossRef]
- Generalovic, T.N.; McCarthy, S.A.; Warren, I.A.; Wood, J.M.D.; Torrance, J.; Sims, Y.; Quail, M.; Howe, K.; Pipan, M.; Durbin, R.; et al. A high-quality, chromosome-level genome assembly of the Black Soldier Fly (Hermetia illucens L.). G3 Genes|Genomes|Genetics 2021, 11, jkab085. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Sherrill-Mix, S. Package ‘Taxonomizr’. Available online: https://cran.r-project.org/web/packages/taxonomizr/ (accessed on 26 January 2021).
- Dowle, M.; Short, T.; Lianoglou, S.; Srinivasan, A. R: Data.Table. Available online: http://r-datatable.com (accessed on 26 January 2021).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 27 March 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 2019, 8, giz100. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2020, 39, 578–585. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Wickner, R.B.; Ghabrial, S.A.; Nibert, M.L.; Patterson, J.L.; Wang, C.C. Family: Totiviridae. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/dsrna-viruses-2011/w/dsrna_viruses/191/totiviridae (accessed on 4 February 2022).
- Bruenn, J.A. A closely related group of RNA-dependent RNA polymerases from double-stranded RNA viruses. Nucleic Acids Res. 1993, 21, 5667. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Martinez, J.; Lepetit, D.; Ravallec, M.; Fleury, F.; Varaldi, J. Additional heritable virus in the parasitic wasp Leptopilina boulardi: Prevalence, transmission and phenotypic effects. J. Gen. Virol. 2016, 97, 523–535. [Google Scholar] [CrossRef]
- Lester, P.J.; Buick, K.H.; Baty, J.W.; Felden, A.; Haywood, J. Different bacterial and viral pathogens trigger distinct immune responses in a globally invasive ant. Sci. Rep. 2019, 9, 5780. [Google Scholar] [CrossRef]
- Baty, J.W.; Bulgarella, M.; Dobelmann, J.; Felden, A.; Lester, P.J. Viruses and their effects in ants (Hymenoptera: Formicidae). Myrmecol. News 2020, 30, 213–228. [Google Scholar] [CrossRef]
- Nebbak, A.; Monteil-Bouchard, S.; Berenger, J.M.; Almeras, L.; Parola, P.; Desnues, C. Virome diversity among mosquito populations in a sub-urban region of Marseille, France. Viruses 2021, 13, 768. [Google Scholar] [CrossRef]
- Zhai, Y.; Attoui, H.; Jaafar, F.M.; Wang, H.Q.; Cao, Y.X.; Fan, S.P.; Sun, Y.X.; Liu, L.D.; Mertens, P.P.C.; Meng, W.S.; et al. Isolation and full-length sequence analysis of Armigeres subalbatus totivirus, the first totivirus isolate from mosquitoes representing a proposed novel genus (Artivirus) of the family Totiviridae. J. Gen. Virol. 2010, 91, 2836–2845. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Y.; Ge, X.; Yuan, J.; Shi, Z. A novel totivirus-like virus isolated from bat guano. Arch. Virol. 2012, 157, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Guo, X.; Zhang, S.; Zhao, Q.; Sun, Q.; Zhou, H.; Zhang, J.; Tong, Y. Discovery of two novel totiviruses from Culex tritaeniorhynchus classifiable in a distinct clade with arthropod-infecting viruses within the family Totiviridae. Arch. Virol. 2018, 163, 2899–2902. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Du, J.; Wu, Z.; Zhang, W.; Fu, S.; Song, J.; Wang, Q.; He, Y.; Lei, W.; Xu, S.; et al. Identification and genetic analysis of a totivirus isolated from the Culex tritaeniorhynchus in northern China. Arch. Microbiol. 2020, 202, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Fauver, J.R.; Grubaugh, N.D.; Krajacich, B.J.; Weger-Lucarelli, J.; Lakin, S.M.; Fakoli, L.S.; Bolay, F.K.; Diclaro, J.W.; Dabiré, K.R.; Foy, B.D.; et al. West African Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses. Virology 2016, 498, 288–299. [Google Scholar] [CrossRef]
- Katzourakis, A.; Gifford, R.J. Endogenous Viral Elements in Animal Genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef] [PubMed]
- YW Iwasaki, M.S.H.S. PIWI-interacting RNA: Its biogenesis and functions. Annu. Rev. Biochem. 2015, 84, 405–433. [Google Scholar] [CrossRef]
- Drezen, J.M.; Bézier, A.; Burke, G.R.; Strand, M.R. Bracoviruses, ichnoviruses, and virus-like particles from parasitoid wasps retain many features of their virus ancestors. Curr. Opin. Insect Sci. 2022, 49, 93–100. [Google Scholar] [CrossRef]
- Anderson, R.M.; Ay, R.M.M. The population dynamics of microparasites and their invertebrate hosts. Philos. Trans. R. Soc. London. B Biol. Sci. 1981, 291, 451–524. [Google Scholar] [CrossRef]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef]
- Gebremedhn, H.; Deboutte, W.; Schoonvaere, K.; Demaeght, P.; De Smet, L.; Amssalu, B.; Matthijnssens, J.; De Graaf, D.C. Metagenomic approach with the NetoVIR enrichment protocol reveals virus diversity within ethiopian honey bees (Apis mellifera simensis). Viruses 2020, 12, 1218. [Google Scholar] [CrossRef]
- Obbard, D.J. Expansion of the metazoan virosphere: Progress, pitfalls, and prospects. Curr. Opin. Virol. 2018, 31, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Emerman, M.; Malik, H.S. Paleovirology—Modern consequences of ancient viruses. PLoS Biol. 2010, 8, e1000301. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Ferreira, R.; de Toni Aquino da Cruz, L.C.; de Souza, V.J.; da Silva Neves, N.A.; de Souza, V.C.; Filho, L.C.F.; da Silva Lemos, P.; de Lima, C.P.S.; Naveca, F.G.; Atanaka, M.; et al. Insect-specific viruses and arboviruses in adult male culicids from Midwestern Brazil. Infect. Genet. Evol. 2020, 85, 104561. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.T.; Maertens, B.L.; Dunham, T.J.; Rodgers, C.P.; Brehm, A.L.; Miller, M.R.; Williams, A.M.; Foy, B.D.; Stenglein, M.D. Partitiviruses Infecting Drosophila melanogaster and Aedes aegypti Exhibit Efficient Biparental Vertical Transmission. J. Virol. 2020, 94, e01070-20. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Dietzgen, R.G.; Joubert, D.A.; Blasdell, K.R. Rhabdovirus accessory genes. Virus Res. 2011, 162, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Faizah, A.N.; Kobayashi, D.; Isawa, H.; Amoa-Bosompem, M.; Murota, K.; Higa, Y.; Futami, K.; Shimada, S.; Kim, K.S.; Itokawa, K.; et al. Deciphering the virome of Culex vishnui subgroup mosquitoes, the major vectors of japanese encephalitis, in Japan. Viruses 2020, 12, 264. [Google Scholar] [CrossRef]
- Brinton, M.A.; Gulyaeva, A.A.; Balasuriya, U.B.R.; Dunowska, M.; Faaberg, K.S.; Goldberg, T.; Leung, F.C.C.; Nauwynck, H.J.; Snijder, E.J.; Stadejek, T.; et al. ICTV virus taxonomy profile: Arteriviridae 2021. J. Gen. Virol. 2021, 102, 001632. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Family | EVE | BGA | Best Viral Hit | Viral Hit Similarity | Coordinates on BGA Contigs § | ||
|---|---|---|---|---|---|---|---|
| Name | Location † | AA (%) ¤ | Protein # | ||||
| Partitiviridae | PartitiEVE | PT | 1 | Atrato Partiti-like virus 2 | 44.6 | Capsid | JXPW01014295.1:343-1512 |
| np* | 1 | 49 | Capsid | JXPW01121853.1:492-1707 | |||
| np* | 2 | 47.5 | Capsid | VFFH01000694.1:2871386-2872788 | |||
| PT | 2 | 54.8 | Capsid | VFFH01002716.1:5535-6390 | |||
| PT | 3 | 54.1 | Capsid | LR899010.1:144460311-144461333 | |||
| Parvoviridae | ParvoEVE | PR1 | 2 | Clinch densovirus 1 | 66.7 | Capsid | VFFH01002420.1:17403-17642 |
| PR1 | 3 | Densovirinae sp. | 39.2 | Capsid | LR899010.1:21909312-21909550 | ||
| PR2 | 1 | Haematobia irritans densovirus | 45.3 | Capsid | JXPW01295709.1:732-1063 | ||
| np* | 2 | 62.5 | ORF1 | VFFH01002716.1:27731-27993 | |||
| np* | 2 | 33.5 | Capsid | VFFH01002716.1:22618-23330 | |||
| PR2 | 3 | 45.4 | Capsid | LR899010.1:144484849-144485180 | |||
| PR3 | 3 | Lone star tick densovirus 1 | 45.8 | ORF1 | LR899014.1:3976151-3976299 | ||
| Rhabdoviridae | RhabdoEVE | Rh | 2 | Entomophthora rhabdovirus A | 55.1 | RdRP | VFFH01000694.1:2885224-2885413 |
| Totiviridae | TotiEVE | T1 | 1 | Leptopilina boulardi toti-like virus | 54.8 | RdRP | JXPW01175605.1:2029-3362 |
| T2 | 1 | 34.6 | Capsid | JXPW01052892.1:5735-9302 | |||
| T1 | 1 | 28.6 | Capsid | JXPW01318472.1:69-1591 | |||
| T3 | 1 | 33.2 | Capsid | JXPW01168285.1:326-1578 | |||
| T1 | 2 | 53 | RdRP | VFFH01002277.1:524489-528239 | |||
| T2 | 2 | 30.4 | Capsid | VFFH01001437.1:1443067-1446431 | |||
| np* | 2 | 38.5 | Capsid | VFFH01001390.1:32171-33459 | |||
| T3 | 2 | 36.7 | Capsid | VFFH01001777.1:322470-323680 | |||
| T2 | 3 | 30.4 | Capsid | LR899013.1:31694664-31698028 | |||
| T3 | 3 | 36.8 | Capsid | LR899014.1:10621516-10622714 | |||
| np* | 1 | Linepithema humile toti-like virus 1 | 38.9 | Capsid | JXPW01318876.1:130-1861 | ||
| T1 | 3 | Dumyat virus | 35.4 | RdRP | LR899009.1:97208286-97212028 | ||
| np* | 1 | 39.9 | RdRP | JXPW01237450.1:1885-3346 | |||
| Xinmoviridae | XinmoEVE | Xi | 3 | Lepidopteran anphe-related virus OKIAV50 | 61.6 | RdRP | LR899013.1:111581535-111582826 |
| HiTV1 Contig | SRA Number (Bioproject) | Sample | Reference |
|---|---|---|---|
| contig 1 | SRR14339788 (PRJNA573413) | Midgut of four-day-old larvae reared on food waste | [36] |
| contig 2 | SRR10158821.1 (PRJNA573413) | Midgut of four-day-old larvae reared on cow manure | [36] |
| contig 3 | SRR14339795 (PRJNA573413) | Midgut of eight-day-old larvae reared on cow manure | [36] |
| contig 4 | ERR1801992.1 (PRJEB19091) | Five individual larvae | [33] |
| contig 5 | SRR8242288 (PRJNA506627) | Egg Mass | [35] |
| Name | Transcripts Per Million (TPM) § | ||||
|---|---|---|---|---|---|
| HiTV1 Contig 1 | HiTV1 Contig 2 | HiTV1 Contig 3 | HiTV1 Contig 4 | HiTV1 † Contig 5 | |
| HiTV1 pol | 9343 | 6192 | 3416 | 3590 | 204,488 |
| HiTV1 gag | 6848 | 3473 | 2302 | 2494 | 240,733 |
| Actin-5C | 983,809 | 990,335 | 994,282 | 993,916 | 554,779 |
| Ratio pol/Actin-5C | 0.009 | 0.006 | 0.003 | 0.004 | 0.369 |
| Ratio gag/ Actin-5C | 0.007 | 0.004 | 0.002 | 0.003 | 0.434 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pienaar, R.D.; Gilbert, C.; Belliardo, C.; Herrero, S.; Herniou, E.A. First Evidence of Past and Present Interactions between Viruses and the Black Soldier Fly, Hermetia illucens. Viruses 2022, 14, 1274. https://0-doi-org.brum.beds.ac.uk/10.3390/v14061274
Pienaar RD, Gilbert C, Belliardo C, Herrero S, Herniou EA. First Evidence of Past and Present Interactions between Viruses and the Black Soldier Fly, Hermetia illucens. Viruses. 2022; 14(6):1274. https://0-doi-org.brum.beds.ac.uk/10.3390/v14061274
Chicago/Turabian StylePienaar, Robert D., Clément Gilbert, Carole Belliardo, Salvador Herrero, and Elisabeth A. Herniou. 2022. "First Evidence of Past and Present Interactions between Viruses and the Black Soldier Fly, Hermetia illucens" Viruses 14, no. 6: 1274. https://0-doi-org.brum.beds.ac.uk/10.3390/v14061274