
Ichthyoses—A Clinical and Pathological Spectrum from Heterogeneous Cornification Disorders to Inflammation

Abstract
:1. Target Readership
2. Introduction
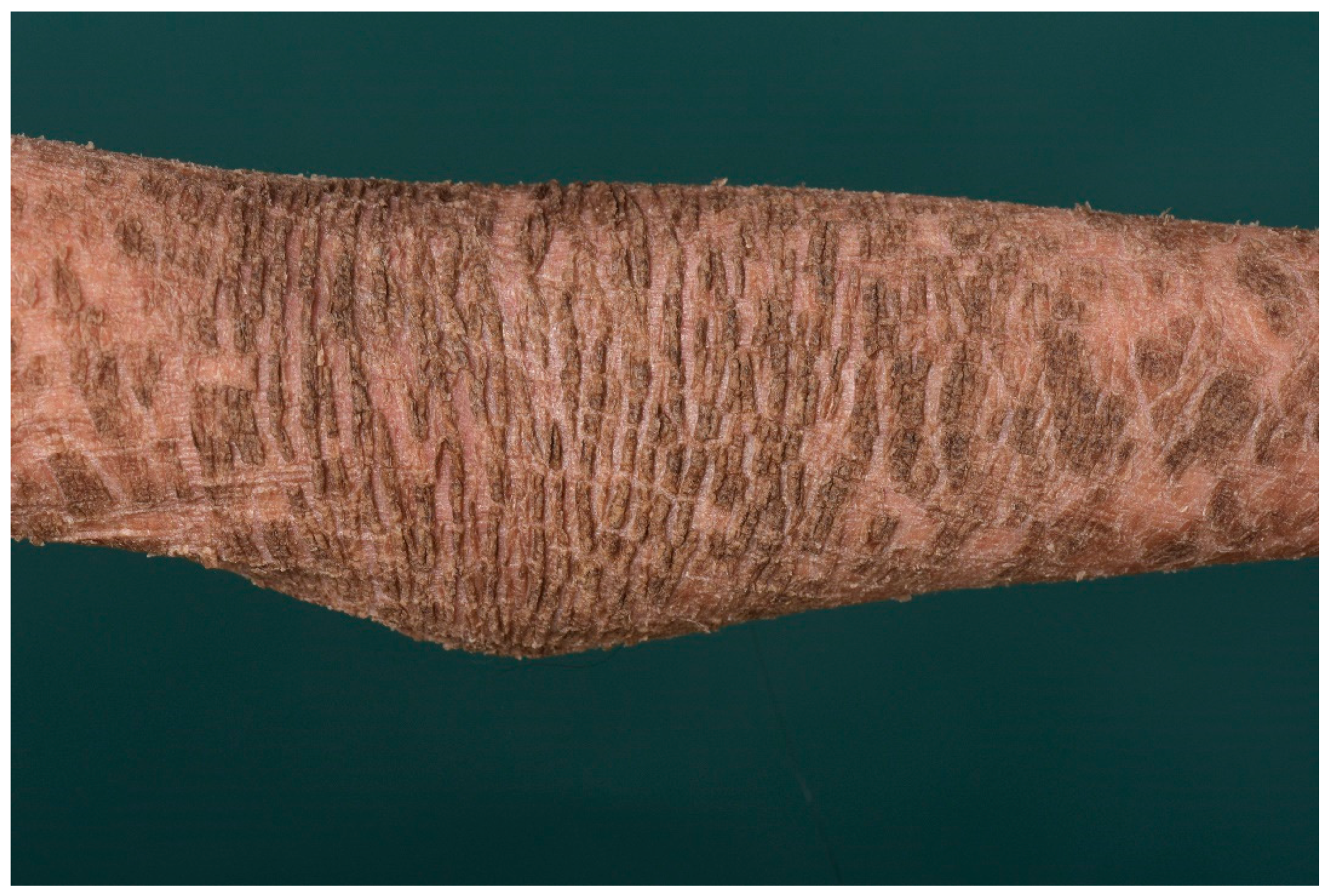
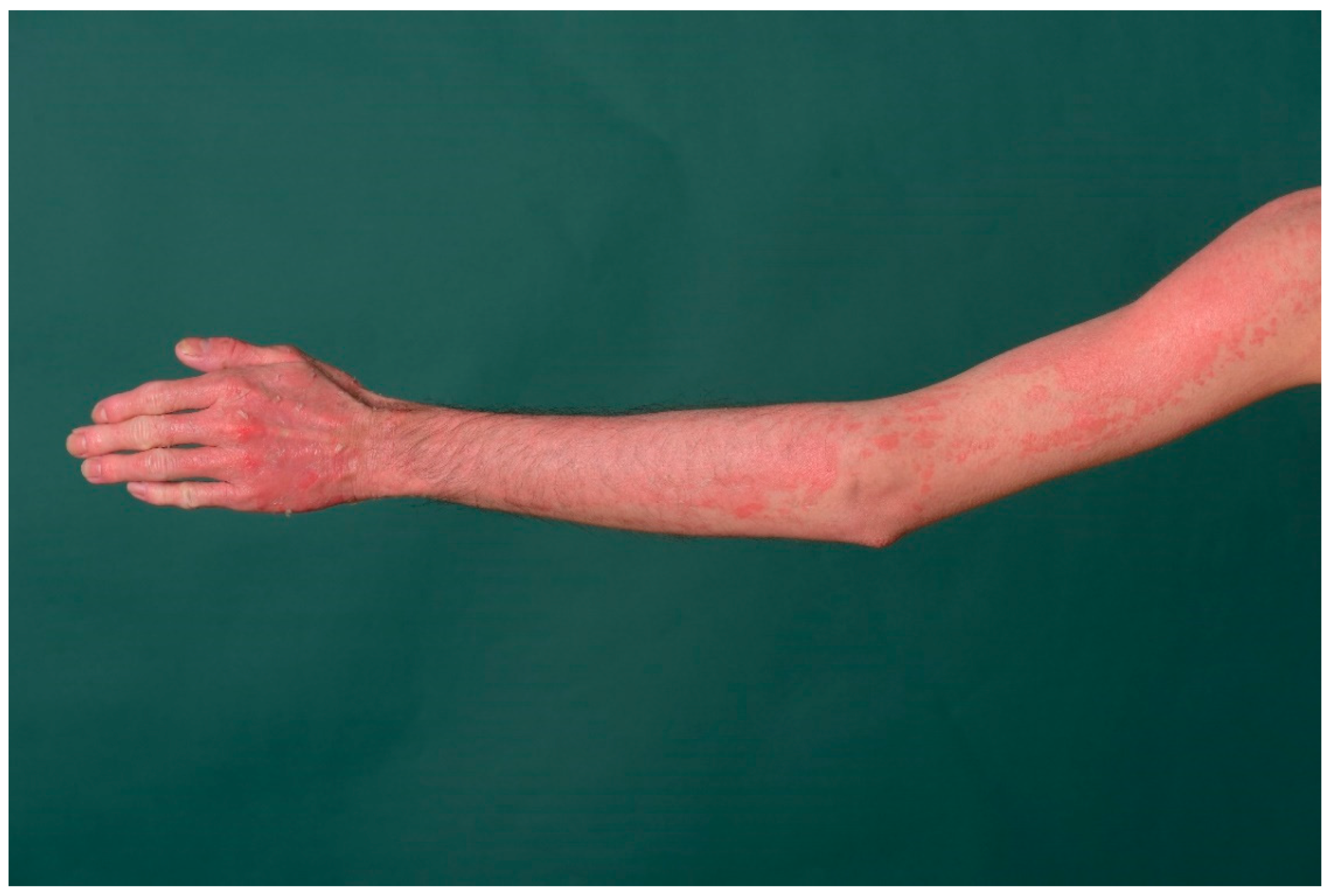
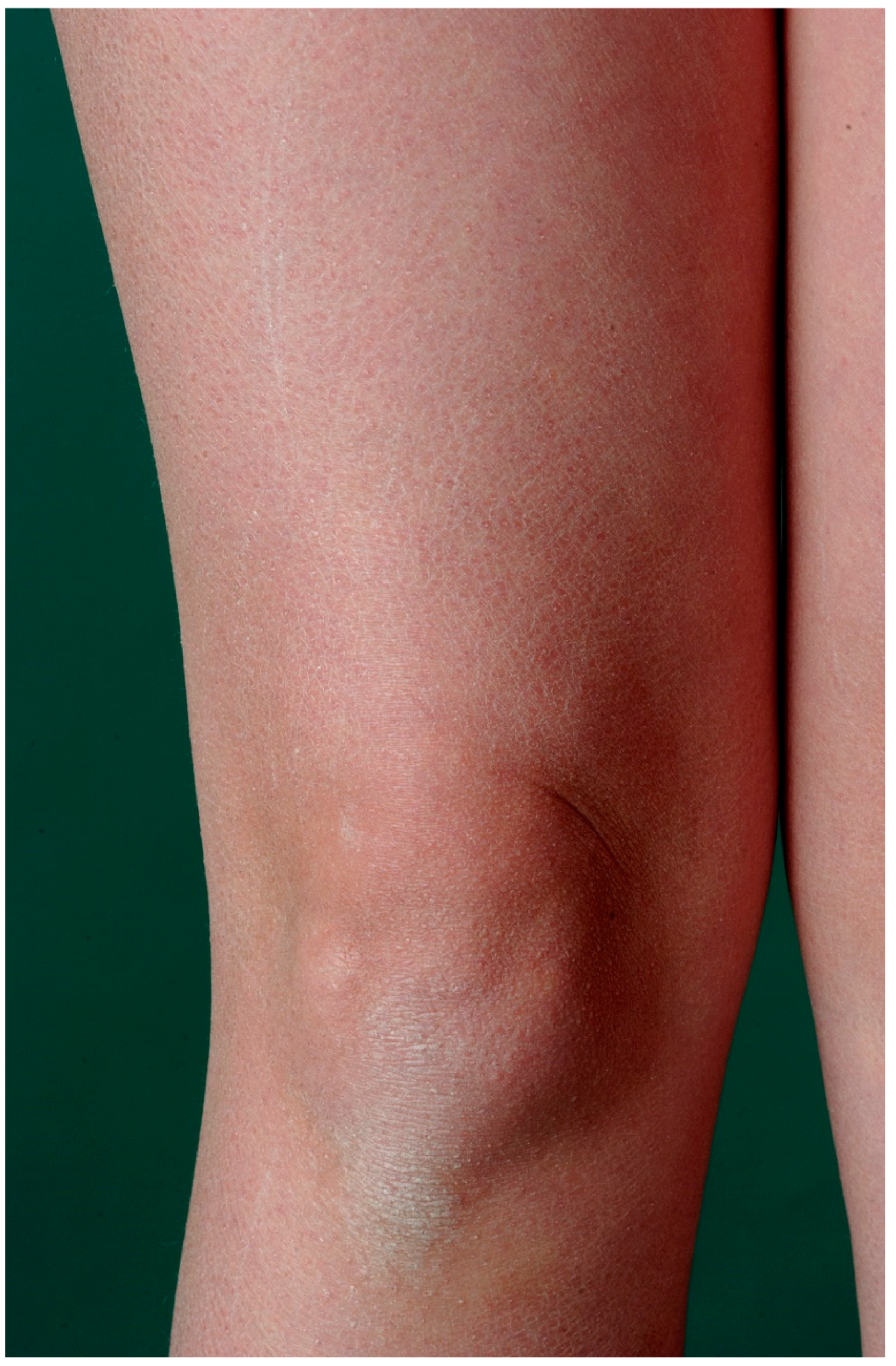
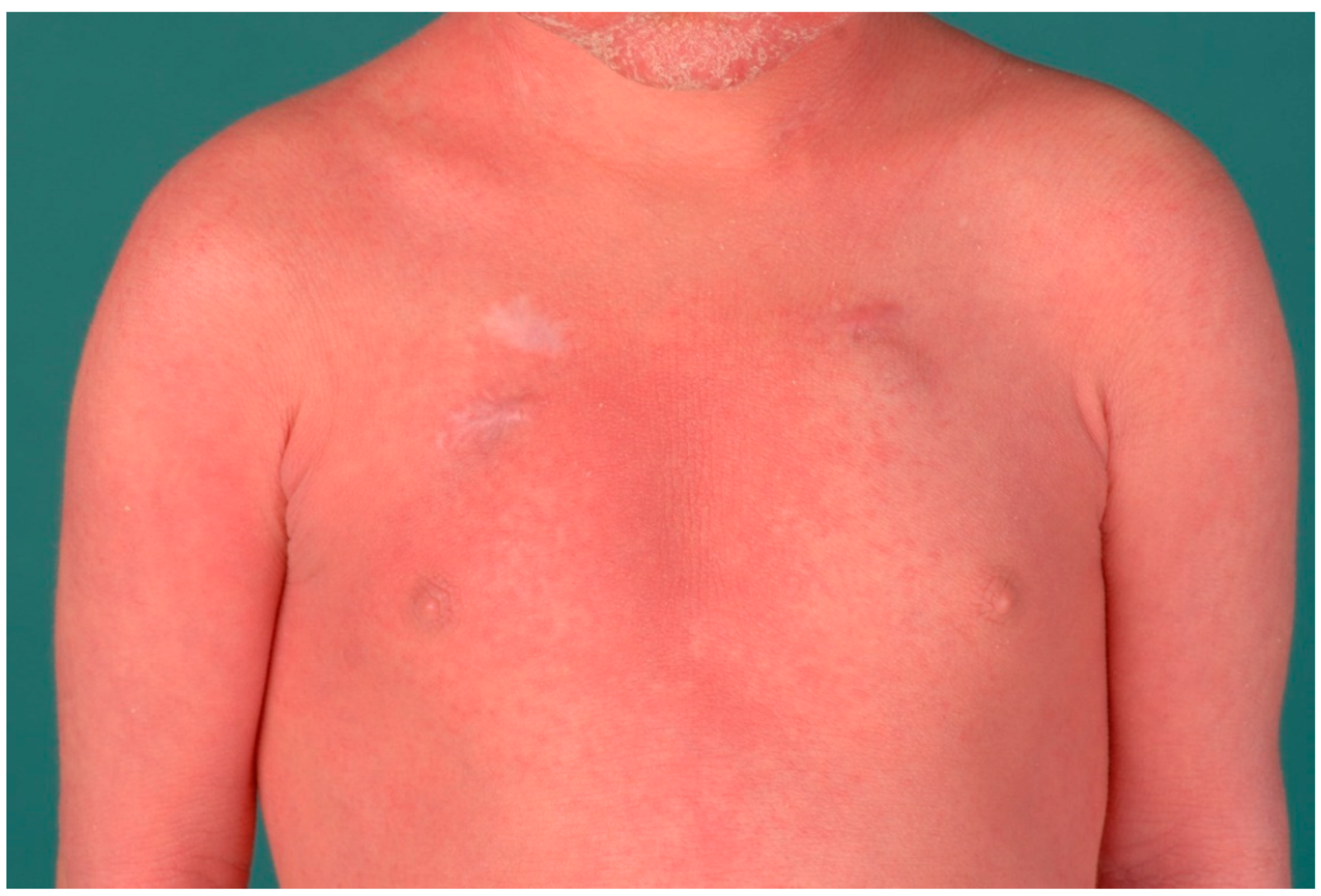
3. Ichthyosis Vulgaris
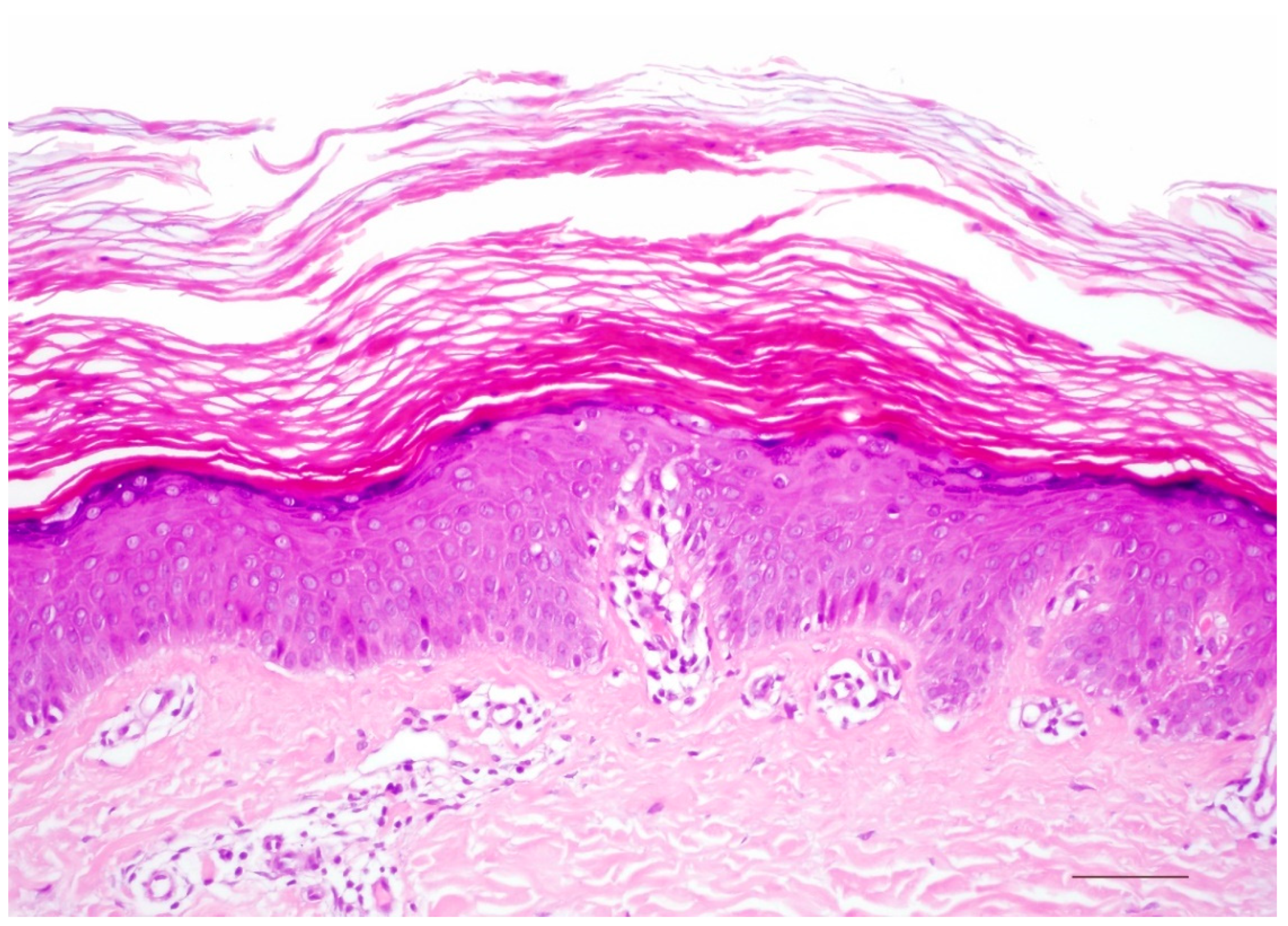
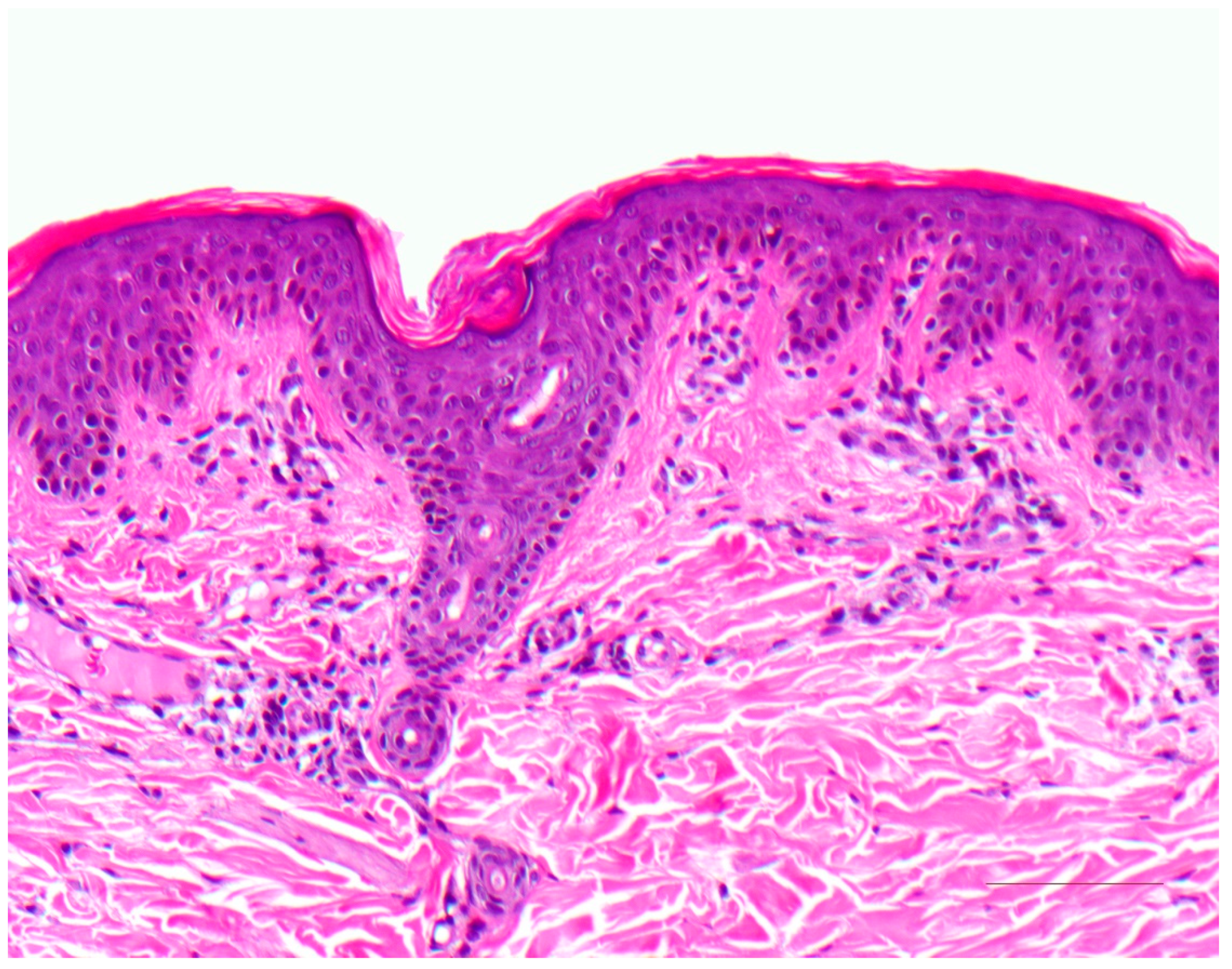
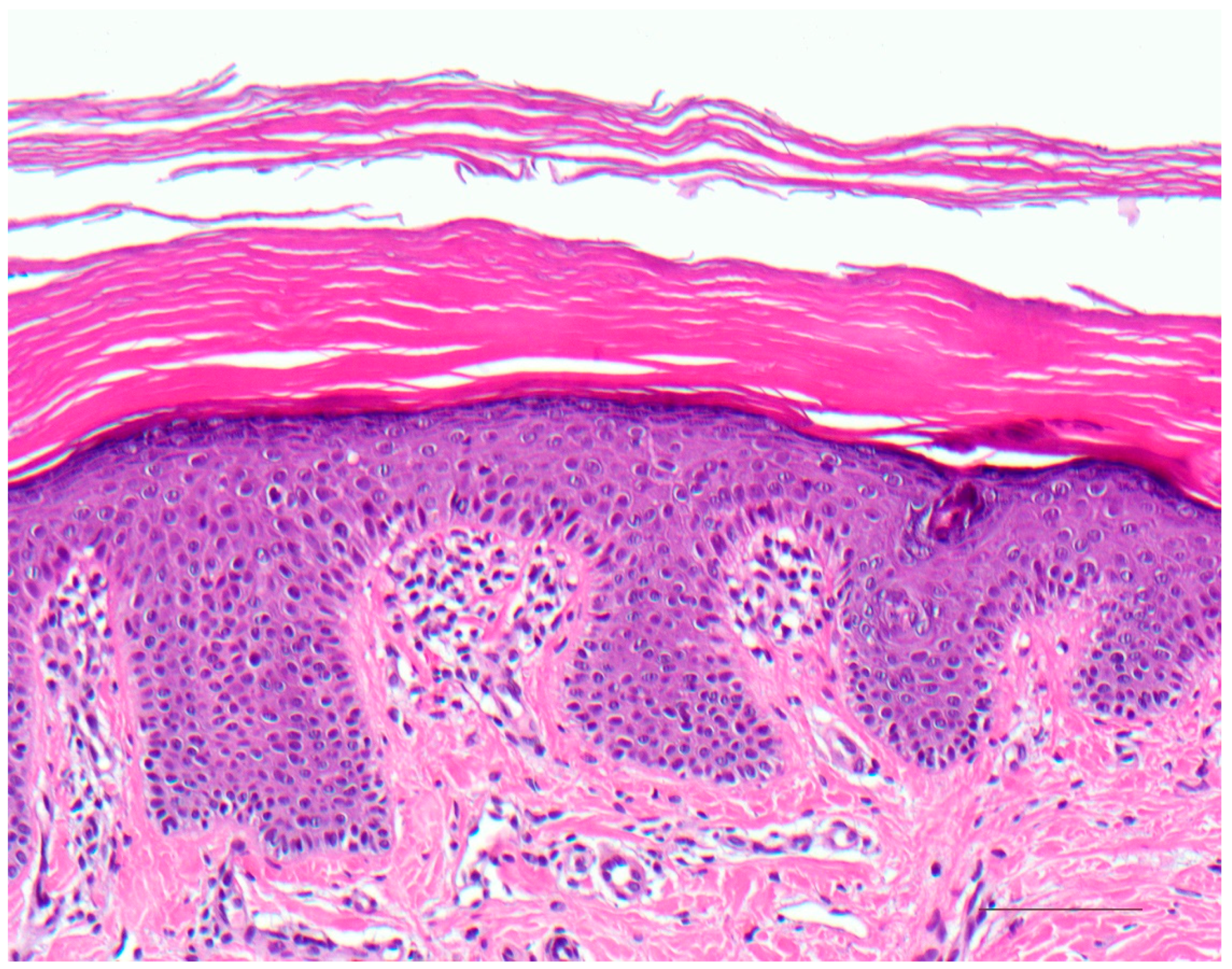
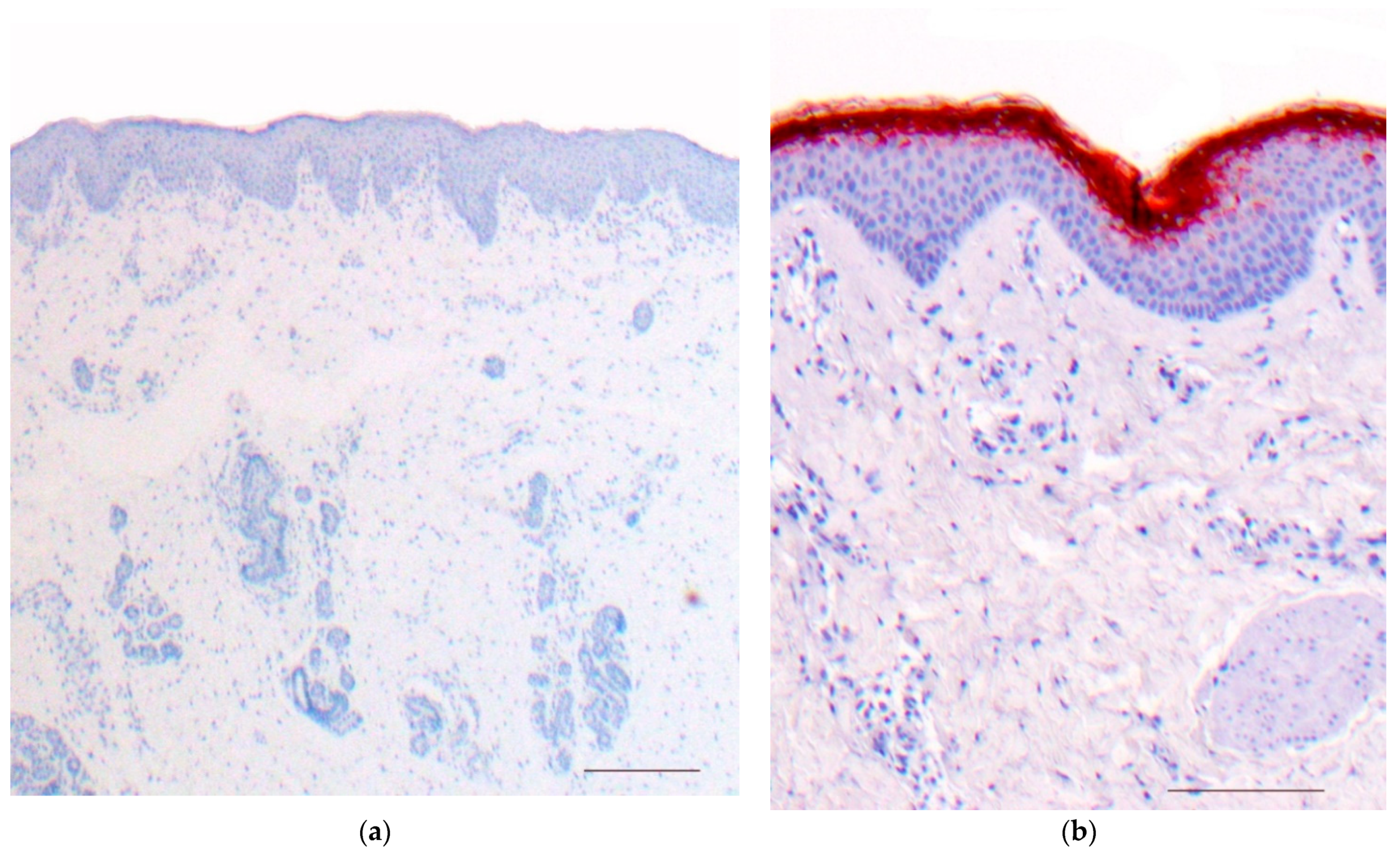
3.1. Histology
3.2. Differential Diagnoses
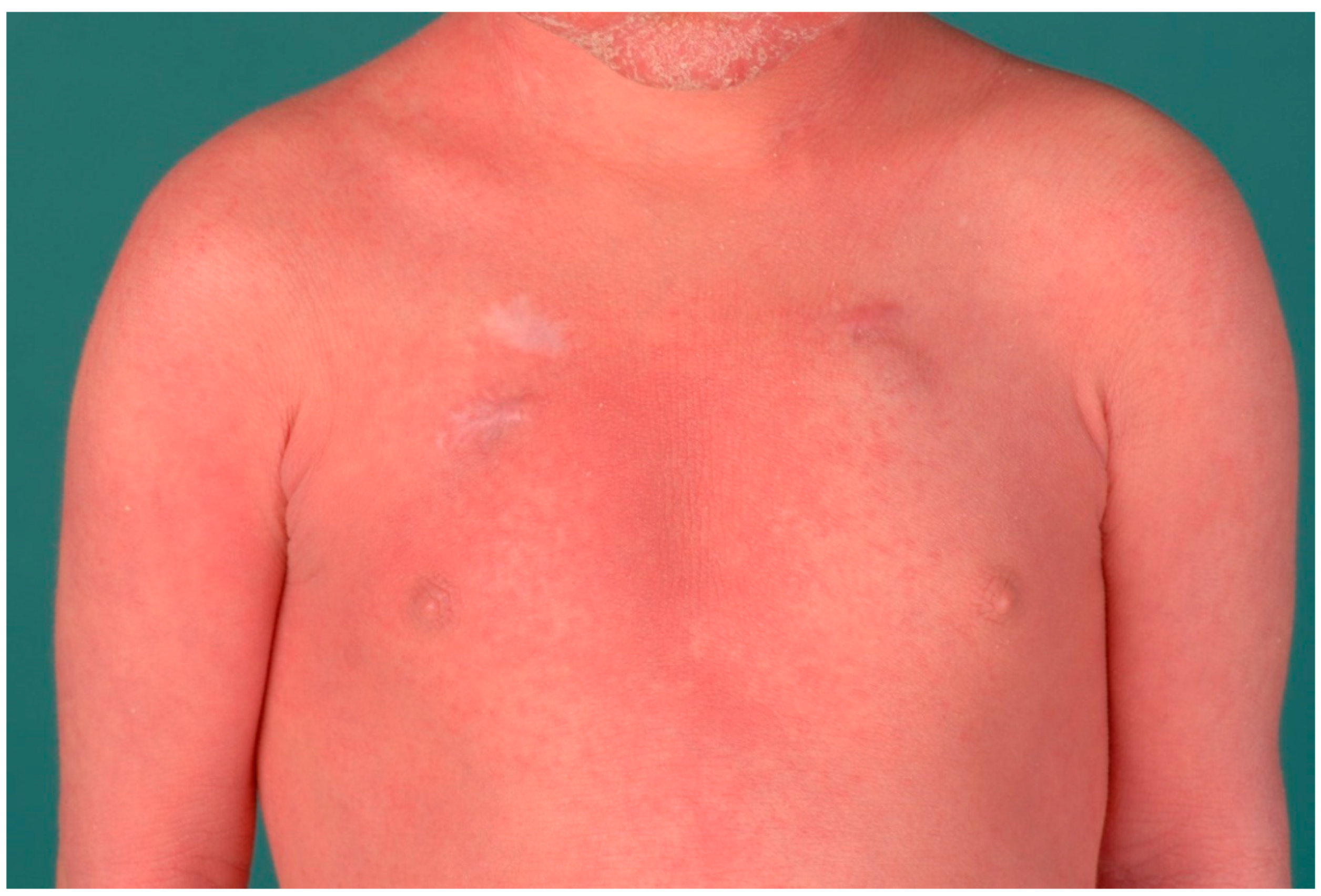
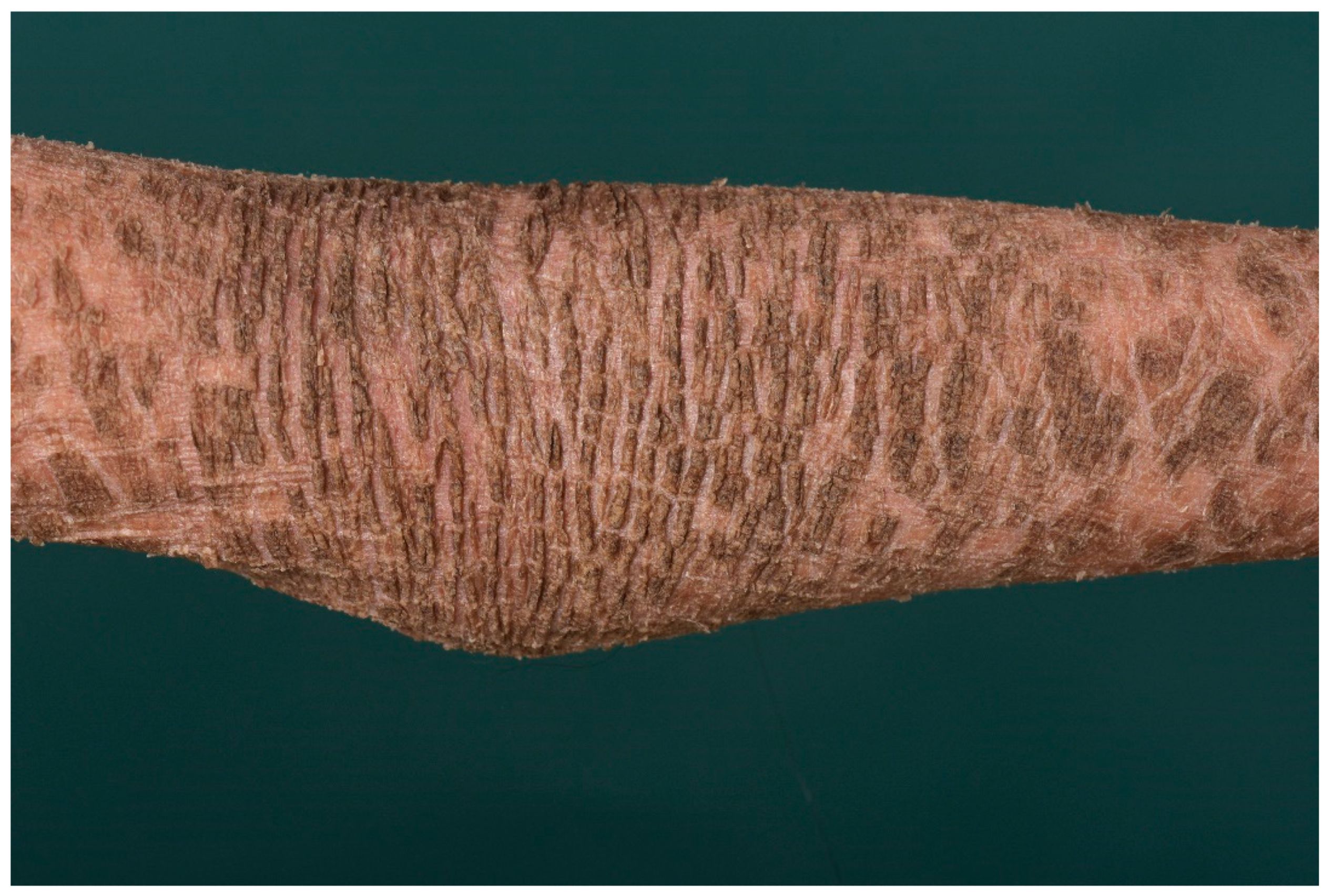
4. Autosomal Recessive Congenital Ichthyosis
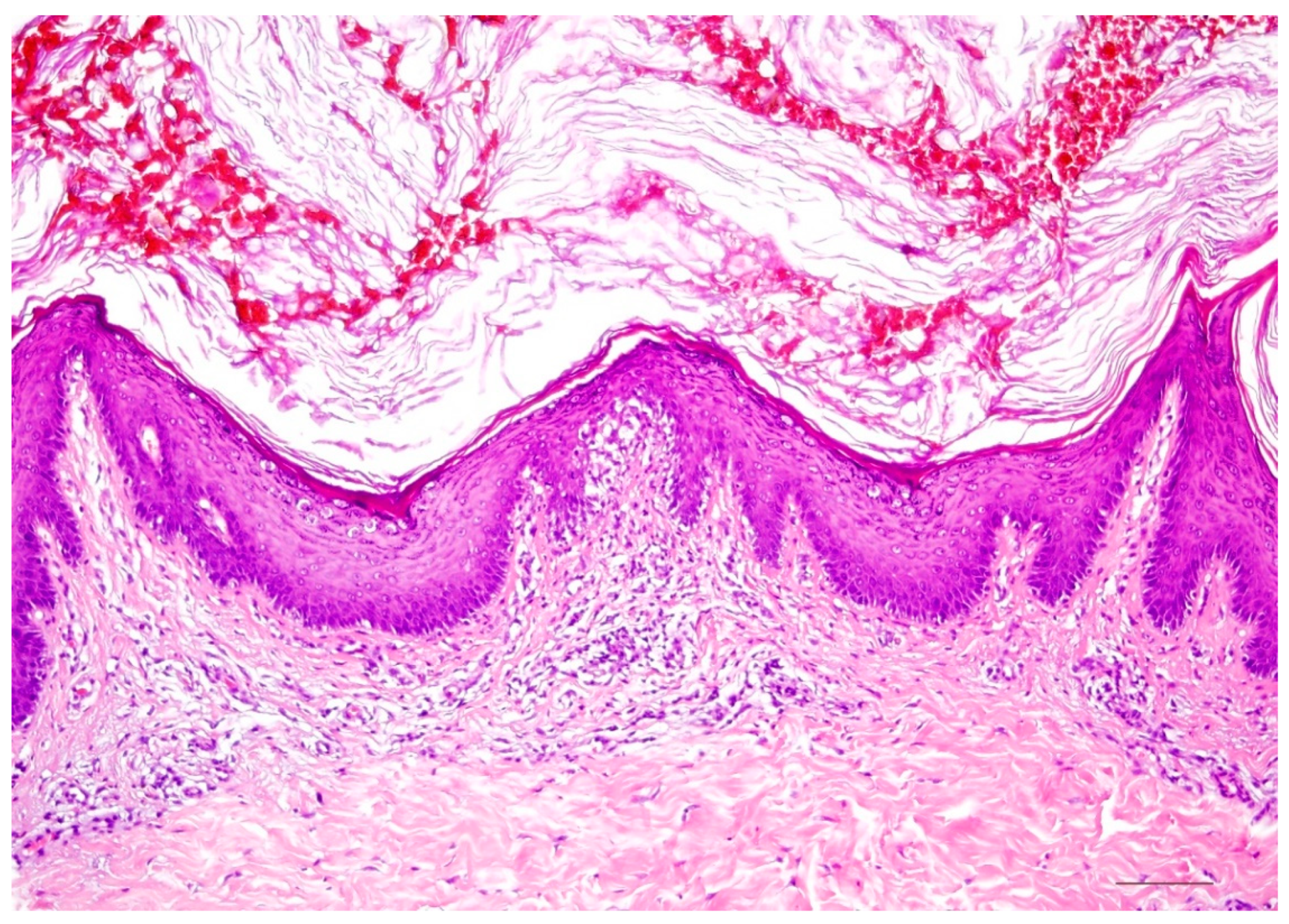
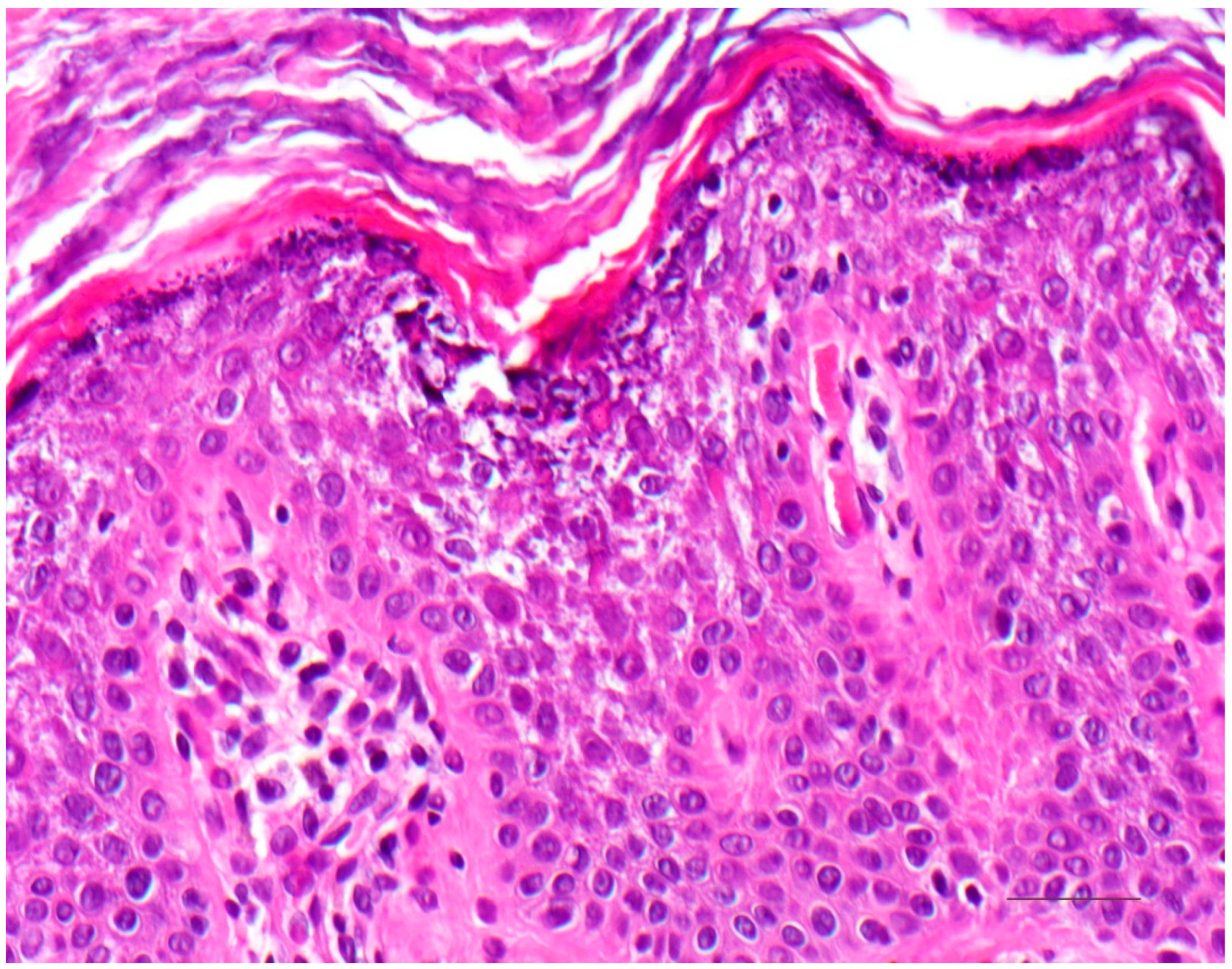
4.1. Histology
4.2. Differential Diagnoses
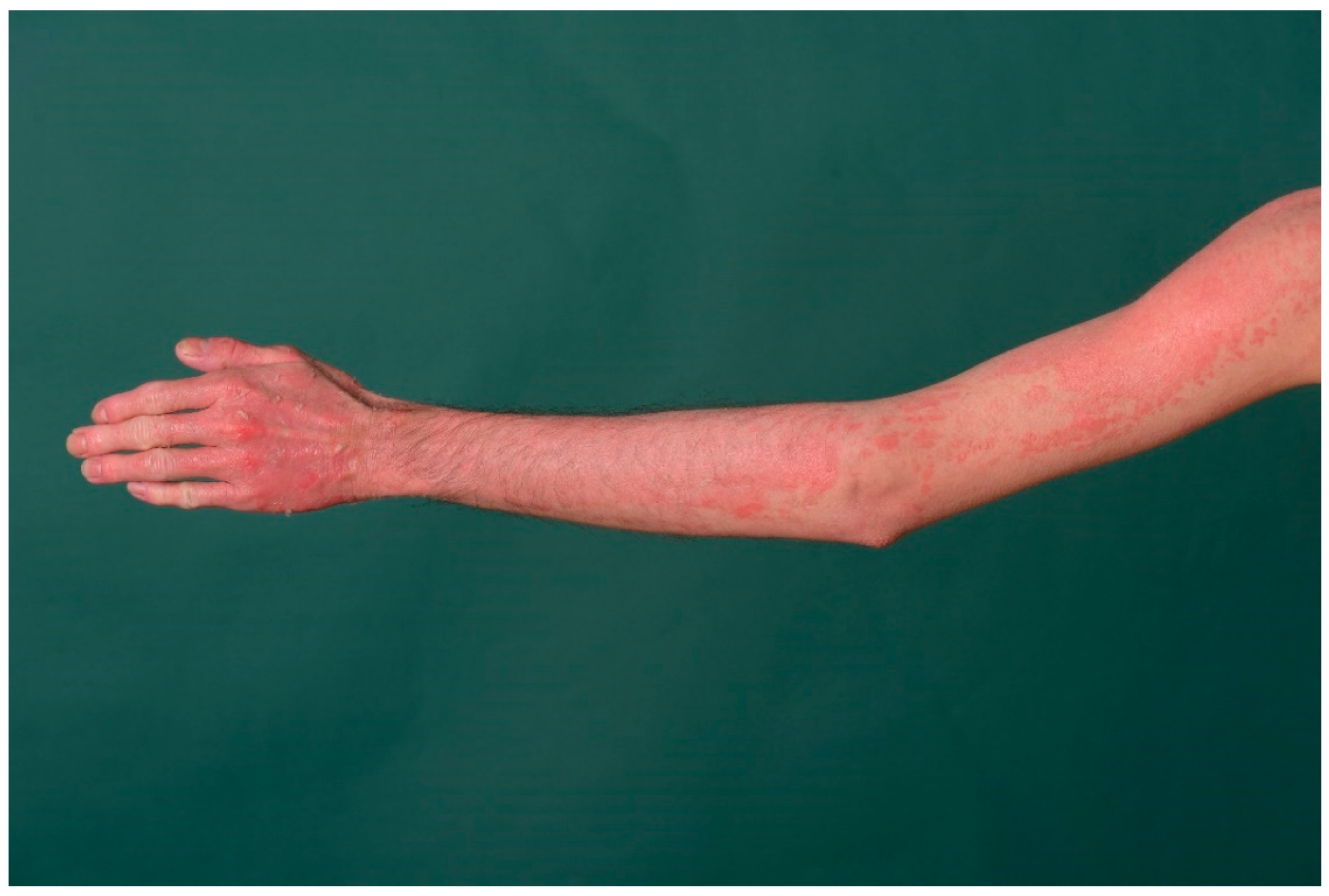
5. Keratinopathic Ichthyosis
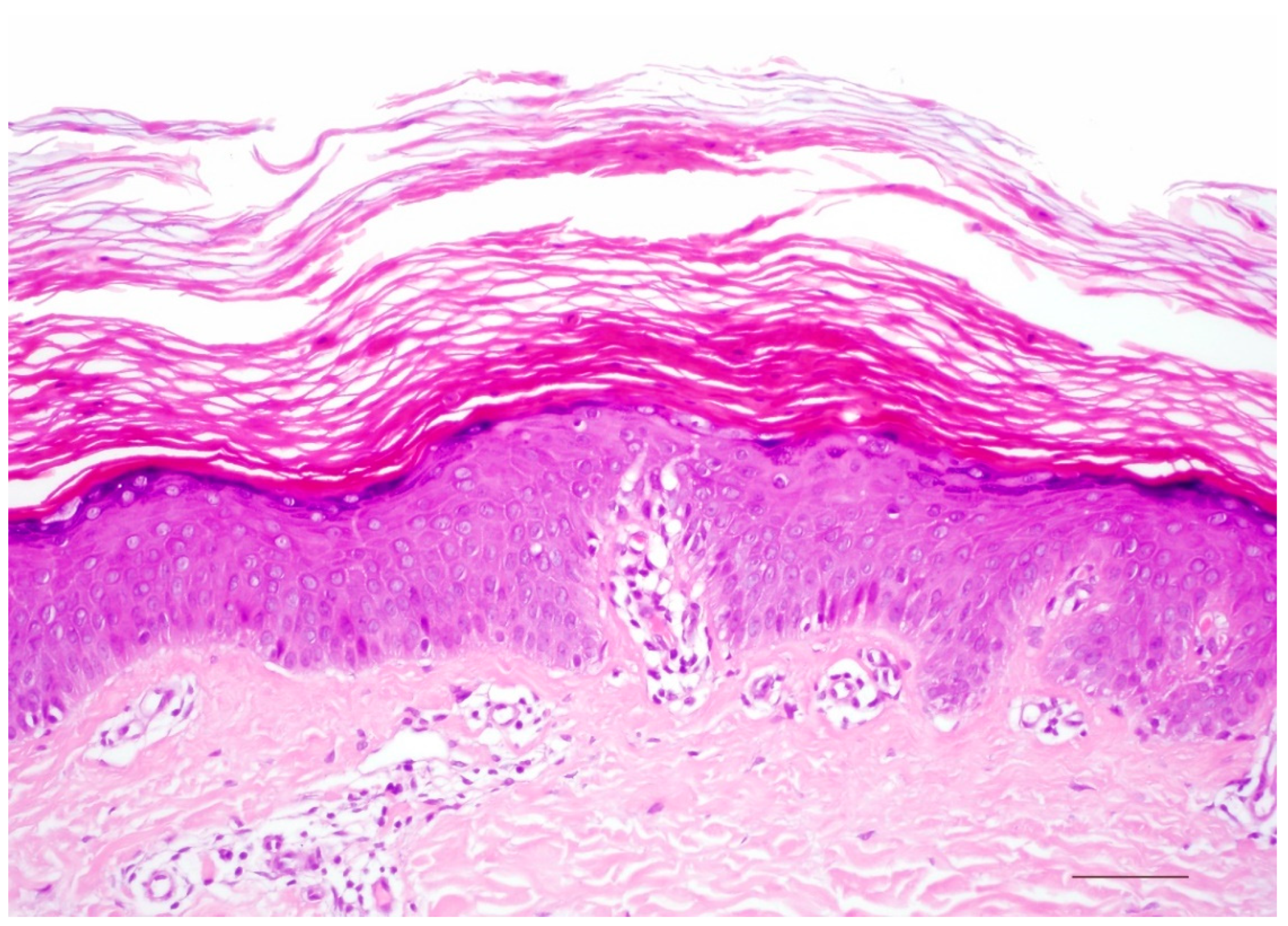
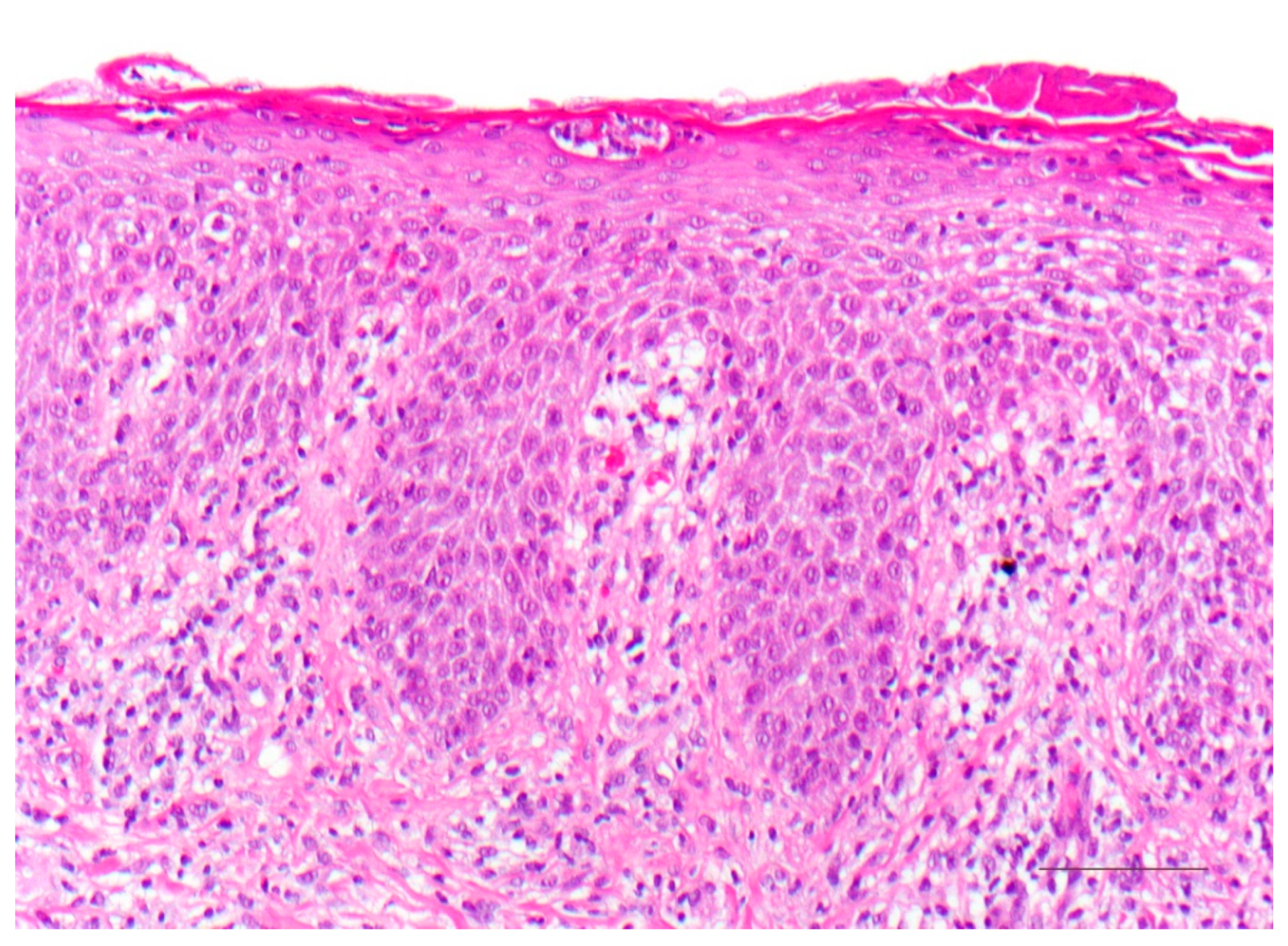
5.1. Histology
5.2. Differential Diagnoses
6. Erythrokeratoderma
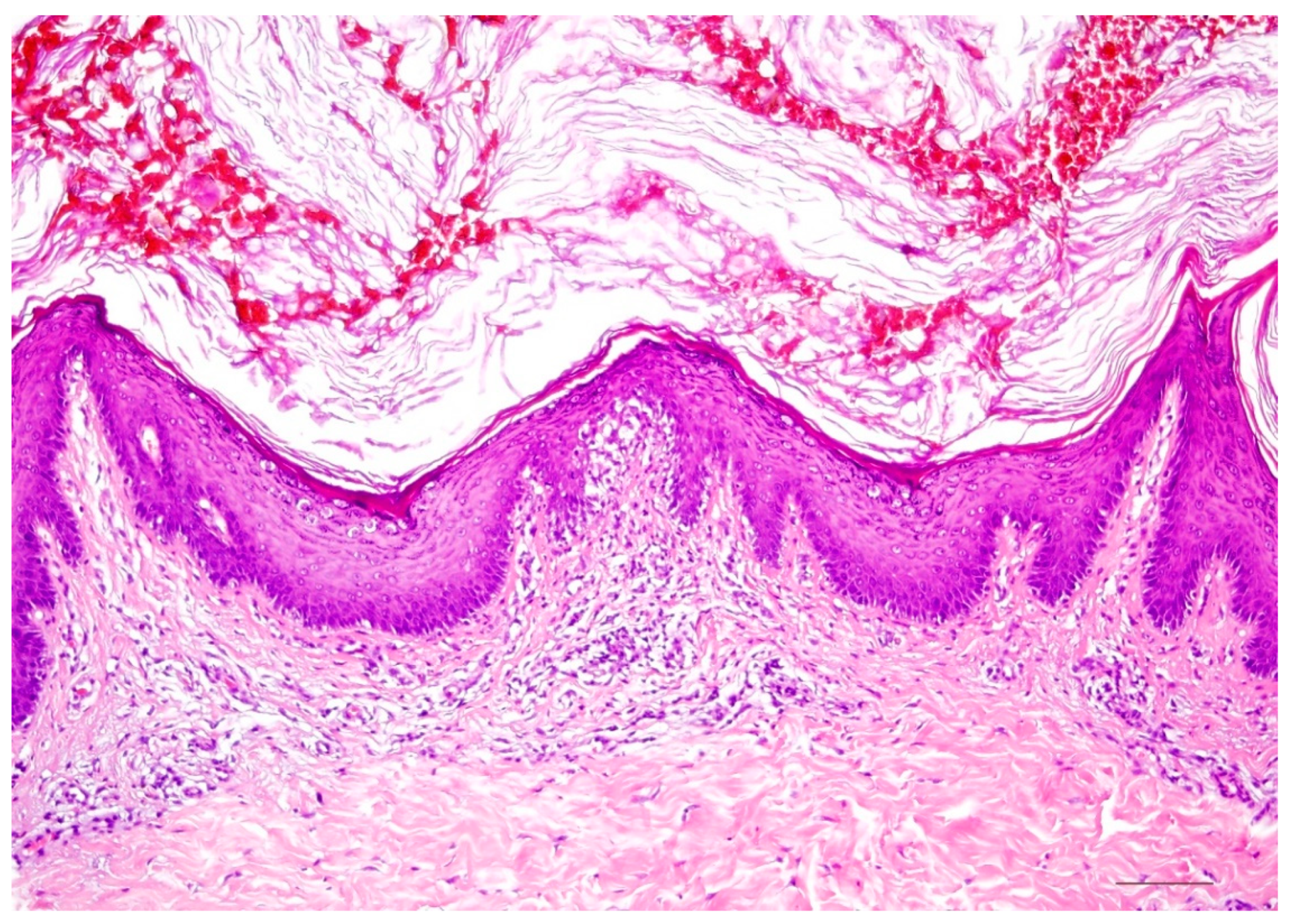
Histology
7. KID Syndrome and HID Syndrome
7.1. Histology
7.2. Differential Diagnoses
8. Ichthyoses with Inflammatory Psoriasiform Pattern
9. Netherton Syndrome
9.1. Histology
9.2. Differential Diagnoses
10. Peeling Skin Disease
10.1. Histology
10.2. Differential Diagnosis
11. CHILD Syndrome
11.1. Histology
11.2. Differential Diagnoses
12. Severe Dermatitis, Multiple Allergies, Metabolic Wasting Syndrome (SAM Syndrome)
12.1. Histology
12.2. Differential Diagnoses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| CHILD syndrome | Congenital hemidysplasia with ichthyosiform erythroderma and limb defects syndrome |
| CRIE | Congenital reticular ichthyosiform erythroderma |
| EI | Epidermolytic ichthyosis |
| KID syndrome | Keratitis ichthyosis deafness syndrome |
| SAM syndrome | Congenital erythroderma–hypotrichosis–recurrent infections–multiple food allergies syndrome |
| SEI | Superficial epidermolytic ichthyosis |
References
- Krug, M.; Oji, V.; Traupe, H.; Berneburg, M. Ichthyoses-Part 1: Differential diagnosis of vulgar ichthyoses and therapeutic options. J. Dtsch. Dermatol. Ges. 2009, 7, 511–519. [Google Scholar] [CrossRef]
- Krug, M.; Oji, V.; Traupe, H.; Berneburg, M. Ichthyoses-Part 2: Congenital ichthyoses. J. Dtsch. Dermatol. Ges. 2009, 7, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Traupe, H. The Ichthyoses. In A Guide to Clinical Diagnosis, Genetic Counseling, and Therapy; Springer: Berlin, Germany, 1989; pp. 103–138. [Google Scholar]
- Oji, V.; Traupe, H. Ichthyoses: Differential diagnosis and molecular genetics. Eur. J. Dermatol. 2006, 16, 349–359. [Google Scholar] [PubMed]
- Oji, V.; Tadini, G.; Akiyama, M.; Blanchet Bardon, C.; Bodemer, C.; Bourrat, E.; Coudiere, P.; DiGiovanna, J.J.; Elias, P.; Fischer, J.; et al. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorèze 2009. J. Am. Acad. Dermatol. 2010, 63, 607–641. [Google Scholar] [CrossRef] [Green Version]
- Metze, D.; Traupe, H. Hereditäre Verhornungsstörungen und epidermale Fehlbildungen. In Histopathologie der Haut, 2nd ed.; Cerroni, L., Garbe, C., Metze, D., Kutzner, H., Kerl, H., Eds.; Springer: Berlin, Germany, 2016; Chapter 20; pp. 404–438. [Google Scholar]
- Metze, D. Disorders of Keratinization. In McKee’s Pathology of the Skin, 5th ed.; 2-Volume-Set; Calonje, E., Brenn, T., Lazar, A., McKee, P.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 1, Chapter 3; pp. 53–117. [Google Scholar]
- Oji, V.; Metze, D.; Traupe, H. Inherited disorders of cornification. In Rook’s Textbook of Dermatology, 9th ed.; Burns, T., Breathnach, S., Cox, N., Griffiths, C., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2016; Volume 2, Part 6; Chapter 65; pp. 1–75. [Google Scholar]
- Majmundar, V.D.; Baxi, K. Hereditary and Acquired Ichthyosis Vulgaris. In Treasure Island (FL); StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Süßmuth, K.; Gruber, R.; Rodriguez, E.; Traupe, H.; Amler, S.; Sánchez-Guijo, A.; Valentin, F.; Tarinski, T.; Straub, N.; Metze, D.; et al. Increased prevalence of filaggrin deficiency in 51 patients with recessive X-linked ichthyosis presenting for dermatologic examination. J. Investig. Dermatol. 2018, 138, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kütting, B.; Traupe, H. Der erworbene Ichthyosis-ähnliche Hautzustand. Hautarztand 1995, 46, 836–840. [Google Scholar] [CrossRef]
- Hotz, A.; Kopp, J.; Bourrat, E.; Oji, V.; Komlosi, K.; Giehl, K.; Bouadjar, B.; Bygum, A.; Tantcheva-Poor, I.; Hellström Pigg, M.; et al. Meta-Analysis of Mutations in ALOX12B or ALOXE3 Identified in a Large Cohort of 224 Patients. Genes 2021, 12, 80. [Google Scholar] [CrossRef]
- Raghunath, M.; Hennies, H.C.; Velten, F.; Wiebe, V.; Steinert, P.M.; Reis, A.; Traupe, H. A novel in situ method for the detection of deficient transglutaminase activity in the skin. Arch. Dermatol. Res. 1998, 290, 621–627. [Google Scholar] [CrossRef]
- Rothnagel, J.A.; Dominey, A.M.; Dempsey, L.D.; Longley, M.A.; Greenhalgh, D.A.; Gagne, T.A.; Huber, M.; Frenk, E.; Hohl, D.; Roop, D.R. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science 1992, 257, 1128–1130. [Google Scholar] [CrossRef]
- Rothnagel, J.A.; Traupe, H.; Wojcik, S.; Huber, M.; Hohl, D.; Pittelkow, M.R.; Saeki, H.; Ishibashi, Y.; Roop, D.R. Mutations in the rod domain of keratin 2e in patients with ichthyosis bullosa of Siemens. Nat. Genet. 1994, 7, 485–490. [Google Scholar] [CrossRef]
- Traupe, H.; Kolde, G.; Hamm, H.; Happle, R. Ichthyosis bullosa of Siemens: A unique type of epidermolytic hyperkeratosis. J. Am. Acad. Dermatol. 1986, 14, 1000–1005. [Google Scholar] [CrossRef]
- Avshalumova, L.; Fabrikant, J.; Koriakos, A. Overview of skin diseases linked to connexin gene mutations. Int. J. Dermatol. 2014, 53, 192–205. [Google Scholar] [CrossRef] [PubMed]
- van Steensel, M.A.M.; Oranje, A.P.; van der Schroeff, J.G.; Wagner, A.; van Geel, M. The missense mutation G12D in connexin30.3 can cause both erythrokeratodermia variabilis of Mendes da Costa and progressive symmetric erythrokeratodermia of Gottron. Am. J. Med. Genet. A 2009, 149A, 657–661. [Google Scholar] [CrossRef]
- van Geel, M.; van Steensel, M.A.; Küster, W.; Hennies, H.C.; Happle, R.; Steijlen, P.M.; König, A. HID and KID syndromes are associated with the same connexin 26 mutation. Br. J. Dermatol. 2002, 146, 938–942. [Google Scholar] [CrossRef]
- Süßmuth, K.; Traupe, H.; Metze, D.; Oji, V. Ichthyoses in everyday practice: Management of a rare group of diseases. J. Dtsch. Dermatol. Ges. 2020, 18, 225–243. [Google Scholar] [CrossRef] [Green Version]
- Chavanas, S.; Bodemer, C.; Rochat, A.; Hamel-Teillac, D.; Ali, M.; Irvine, A.D.; Bonafé, J.L.; Wilkinson, J.; Taïeb, A.; Barrandon, Y.; et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000, 25, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Leclerc-Mercier, S.; Bodemer, C.; Furio, L.; Hadj-Rabia, S.; de Peufeilhoux, L.; Weibel, L.; Bursztejn, A.C.; Bourrat, E.; Ortonne, N.; Molina, T.J.; et al. Skin biopsy in Netherton Syndrome: A Histological review of a large series and new findings. Am. J. Dermatopathol. 2016, 38, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Oji, V.; Eckl, K.M.; Aufenvenne, K.; Nätebus, M.; Tarinski, T.; Ackermann, K.; Seller, N.; Metze, D.; Nürnberg, G.; Fölster-Holst, R.; et al. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: Unraveling the peeling skin disease. Am. J. Hum. Genet. 2010, 87, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramphul, K.; Kota, V.; Mejias, S.G. Child Syndrome. In Treasure Island (FL); StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Happle, R.; Koch, H.; Lenz, W. The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur. J. Pediatr. 1980, 134, 27–33. [Google Scholar] [CrossRef]
- Bergqvist, C.; Abdallah, B.; Hasbani, D.J.; Abbas, O.; Kibbi, A.G.; Hamie, L.; Kurban, M.; Rubeiz, N. CHILD syndrome: A modified pathogenesis-targeted therapeutic approach. Am. J. Med. Genet. A 2018, 176, 733–738. [Google Scholar] [CrossRef]
- König, A.; Happle, R.; Bornholdt, D.; Engel, H.; Grzeschik, K.H. Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am. J. Med. Genet. 2000, 90, 339–346. [Google Scholar] [CrossRef]
- Samuelov, L.; Sarig, O.; Harmon, R.M.; Rapaport, D.; Ishida-Yamamoto, A.; Isakov, O.; Koetsier, J.L.; Gat, A.; Goldberg, I.; Bergman, R.; et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat. Genet. 2013, 45, 1244–1248. [Google Scholar] [CrossRef] [Green Version]
- McAleer, M.A.; Pohler, E.; Smith, F.J.D.; Wilson, N.J.; Cole, C.; MacGowan, S.; Koetsier, J.L.; Godsel, L.M.; Harmon, R.M.; Gruber, R.; et al. Severe dermatitis, multiple allergies, and metabolic wasting syndrome caused by a novel mutation in the N-terminal plakin domain of desmoplakin. J. Allergy. Clin. Immunol. 2015, 136, 1268–1276. [Google Scholar] [CrossRef] [Green Version]
- Taiber, S.; Samuelov, L.; Mohamad, J.; Barak, E.C.; Sarig, O.; Shalev, S.A.; Lestringant, G.; Sprecher, E. SAM syndrome is characterized by extensive phenotypic heterogeneity. Exp. Dermatol. 2018, 27, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Polivka, L.; Hadj-Rabia, S.; Bal, E.; Leclerc-Mercier, S.; Madrange, M.; Hamel, Y.; Bonnet, D.; Mallet, S.; Lepidi, H.; Ovaert, C.; et al. Epithelial barrier dysfunction in desmoglein-1 deficiency. J. Allergy Clin. Immunol. 2018, 142, 702–706.e7. [Google Scholar] [CrossRef] [Green Version]
- Hammers, C.M.; Stanley, J.R. Desmoglein-1, differentiation, and disease. J. Clin. Investig. 2013, 123, 1419–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida-Yamamoto, A.; Igawa, S. Genetic skin diseases related to desmosomes and corneodesmosomes. J. Dermatol. Sci. 2014, 74, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metze, D.; Oji, V. Palmoplantar Keratodermas. In Dermatology, Series: Expert Consult, 4th ed.; Bolognia, J., Schaffer, J., Cerroni, L., Eds.; Elsevier: Philadelphia, PA, USA, 2018; Volume 1, Chapter 58; pp. 924–943. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vulgar ichthyosis, isolated |
| Ichthyosis vulgaris |
| X-linked recessive ichthyosis |
| Vulgar ichthyosis, syndromic |
| Refsum syndrome |
| Multiple sulfatase deficiency |
| Congenital ichthyosis, isolated |
| Keratinopathic ichthyosis |
| Autosomal recessive congenital ichthyosis (ARCI) |
| Harlequin ichthyosis (subtype of ARCI) |
| Autosomal dominant lamellar ichthyosis |
| Congenital reticular ichthyosiform erythroderma (CRIE, Confetti ichthyosis) |
| Ichthyosis hystrix type Curth–Macklin |
| Peeling skin disease |
| Erythrokeratodermia |
| and others |
| Congenital ichthyosis, syndromic |
| HID/KID syndrome |
| Netherton syndrome |
| CHILD syndrome |
| SAM syndrome |
| Conradi–Hünermann–Happle syndrome |
| Sjögren–Larsson syndrome |
| Chanarin–Dorfmann syndrome |
| Trichothiodystrophy |
| IFAP syndrome |
| and others |
| Ichthyoses with Psoriasis-Like Picture |
|---|
| Netherton syndrome |
| Peeling skin disease |
| CHILD syndrome |
| Severe dermatitis, multiple allergies, metabolic wasting syndrome (SAM syndrome) |
| Anular epidermolytic ichthyosis |
| Non-Syndromic Ichthyoses | Common Ichthyoses | Ichthyosis | Gene (mode of inheritance) | |
| Ichthyosis Vulgaris | FLG (filaggrin) (autosomal semidominant) | |||
| X-Linked Ichthyosis | STS (steroid sulfatase) (X-linked recessive) | |||
| ARCI and Keratinopathic Ichthyoses | Harlequin Ichthyosis | ABCA12 (ATP Binding Cassette Subfamily A Member 12) (autosomal recessive) | ||
| Lamellar Ichthyosis, Congenital Ichthyosiform Erythroderma | TGM1 (transglutaminase−1); ALOX12B (Arachidonate 12-Lipoxygenase, 12R Type); ALOXE3 (Arachidonate Lipoxygenase 3); CYP4F22 (Cytochrome P450 Family 4 Subfamily F Member 22); NIPAL4 (Ichthyin) and others (autosomal recessive) | |||
| Bathing Suit Ichthyosis | TGM1 (autosomal recessive) | |||
| Keratinopathic Ichthyoses | EI | KRT1 (keratin 1); KRT10 (keratin 10) (autosomal dominant, sometimes recessive (KRT10 mutations) | ||
| SEI | KRT2 (keratin 2) (autosomal dominant) | |||
| Rare Variants of KPI | CRIE | KRT1 KRT10 (autosomal dominant, de novo mutations) | ||
| Further Non-Syndromic Ichthyoses | Peeling Skin Disease | CDSN (corneodesmosin) (autosomal recessive) | ||
| Erythrokeratoderma Variabilis | GJB3 (encoding Connexin 31) GJB4 (encoding Connexin 30.3) (often autosomal dominant) | |||
| Syndromic Ichthyoses | Netherton Syndrome | SPINK5 (encoding LEKTI) (autosomal recessive) | ||
| KID Syndrome | GJB2 (encoding Connexin 26) (autosomal dominant) | |||
| CHILD Syndrome | NSDHL (NAD(P) Dependent Steroid Dehydrogenase-Like) (x-linked dominant) | |||
| SAM Syndrome | DSG1 (desmoglein−1) DSP (desmoplakin) (autosomal recessive) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metze, D.; Traupe, H.; Süßmuth, K. Ichthyoses—A Clinical and Pathological Spectrum from Heterogeneous Cornification Disorders to Inflammation. Dermatopathology 2021, 8, 107-123. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8020017
Metze D, Traupe H, Süßmuth K. Ichthyoses—A Clinical and Pathological Spectrum from Heterogeneous Cornification Disorders to Inflammation. Dermatopathology. 2021; 8(2):107-123. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8020017
Chicago/Turabian StyleMetze, Dieter, Heiko Traupe, and Kira Süßmuth. 2021. "Ichthyoses—A Clinical and Pathological Spectrum from Heterogeneous Cornification Disorders to Inflammation" Dermatopathology 8, no. 2: 107-123. https://0-doi-org.brum.beds.ac.uk/10.3390/dermatopathology8020017