Hijacking of Lipid Droplets by Hepatitis C, Dengue and Zika Viruses—From Viral Protein Moonlighting to Extracellular Release



Abstract
:1. Introduction
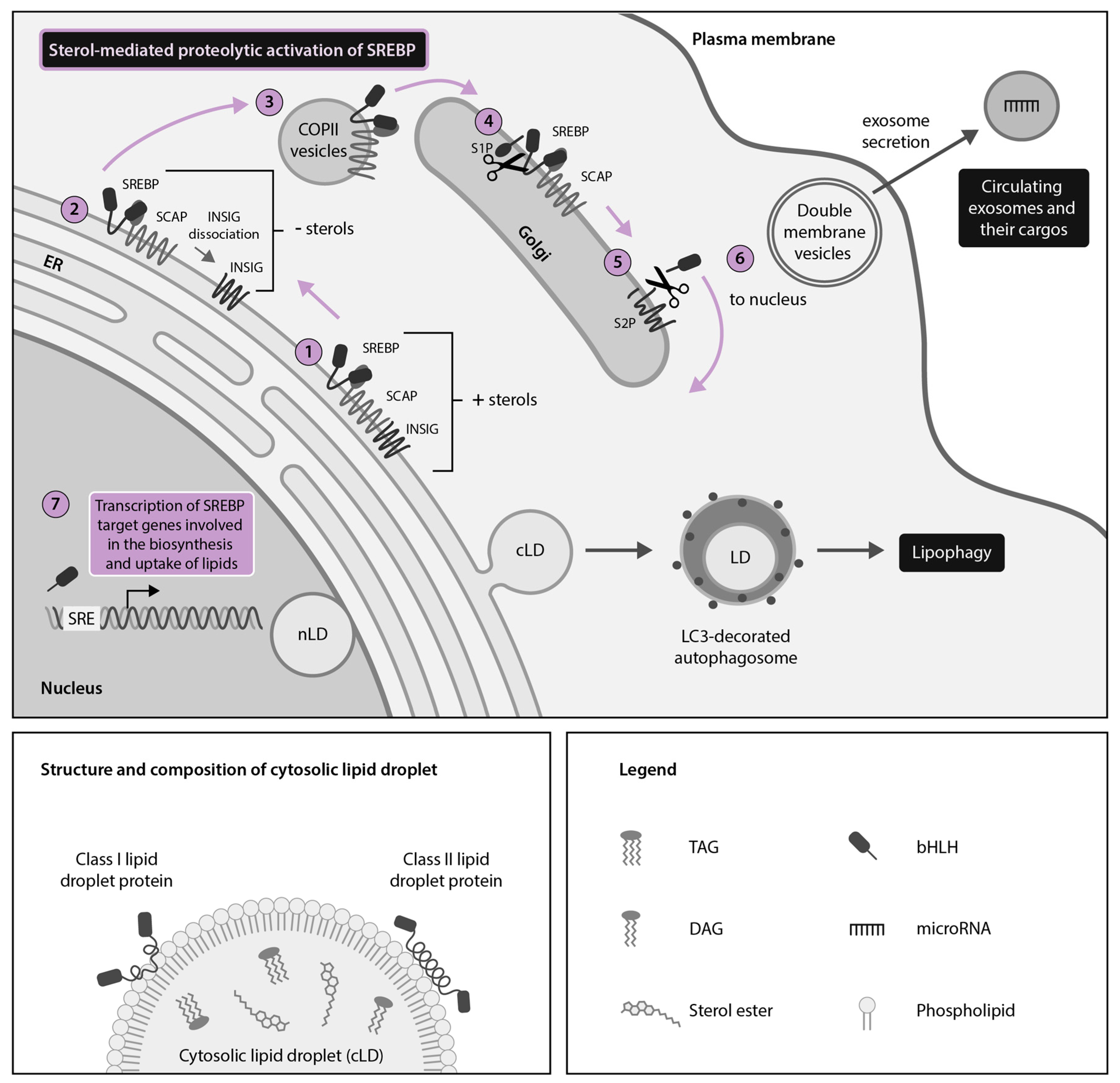
2. HCV, DENV and ZIKV Hijack Cytoplasmic Lipid Droplets (cLDs) Through Dysregulation of the SREBP Pathway
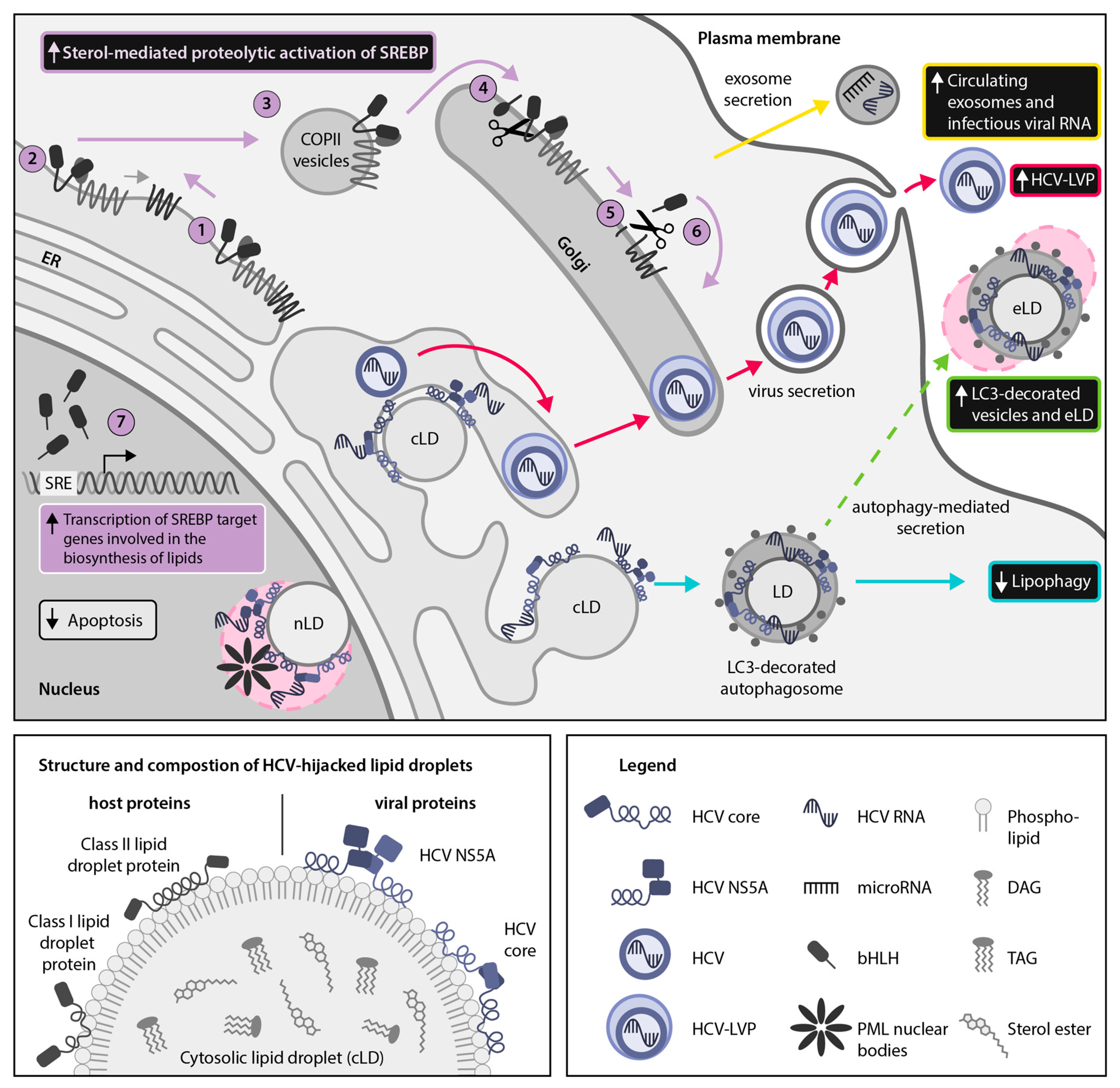
2.1. HCV Usurps the Host SREBP Pathway Through Multiple Mechanisms to Permit the Robust Replication and Production of Infectious Viral Particles
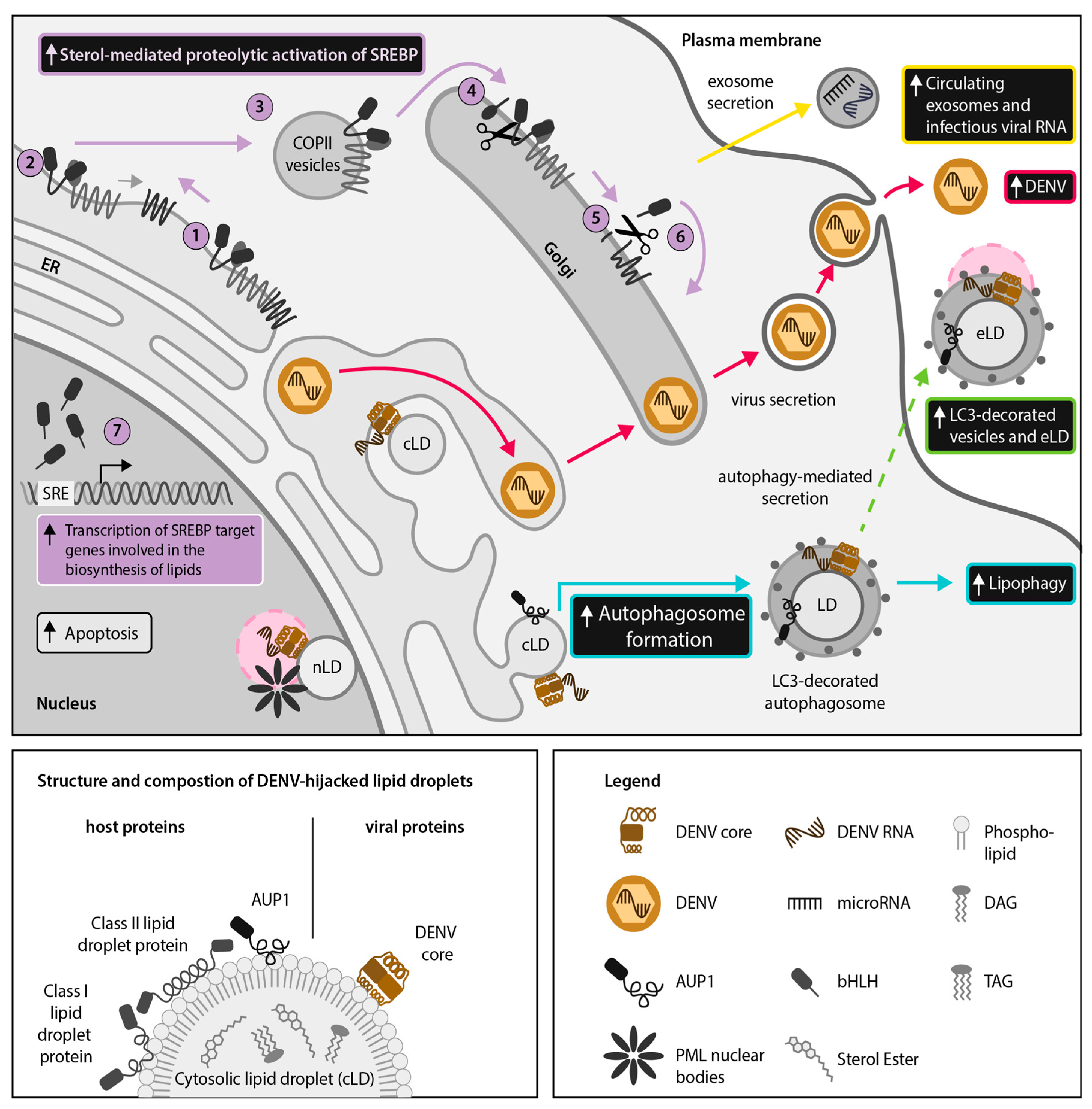
2.2. DENV and ZIKV Divert the SREBP Pathway to Support Their Lifecycle and the Induction of Host Antiviral Responses
3. Discovery of Nuclear LDs (nLDs) and Emerging Moonlighting Activities of LD-Associated Viral Proteins in the Host-Cell Nucleus of Cells Infected With HCV, DENV and ZIKV
4. DENV, ZIKV and HCV Hijack Secretory Autophagy for Viral Dissemination and Release of Extracellular LDs (eLDs) During Infection
4.1. DENV and ZIKV Use Autophagy for Extracellular Transport of LDs and LD-Associated Proteins
4.2. HCV Employs the Secretory Autophagy Pathway for Viral Dissemination
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ago2 AKT AMPK AUP1 Bax | argonaute 2 protein kinase B 5′ adenosine monophosphate-activated protein kinase ancient ubiquitous protein 1 Bcl-2 associated X protein |
| bHLH | basic helix-loop-helix |
| BVDV cLD | bovine viral diarrhea virus cytoplasmic lipid droplet |
| COPII COVID-19 | coat protein II coronavirus disease-2019 |
| CRISPR DAG | clustered regularly interspaced short palindromic repeats diacylglycerol |
| DAXX DDX3X DENV | death domain associated protein DEAD-box helicase 3 X-linked dengue virus |
| eLD | extracellular lipid droplet |
| ER | endoplasmic reticulum |
| HCV | hepatitis C virus |
| HSP IKKα IL-1β JEV INM INSIG | heat shock protein IκB kinase α interleukin-1β Japanese encephalitis virus inner nuclear membrane insulin-induced gene |
| LC3 | microtubule-associated protein light chain 3 |
| LD LDAF1 LDLR | lipid droplet LD assembly factor 1 low-density lipoprotein receptor |
| LVP MERS-CoV miR nLD | Lipoviroparticle Middle East respiratory syndrome coronavirus microRNA nuclear lipid droplet |
| NLS NS | nuclear localization signal non-structural |
| PCSK9 PI3K PML RNAi | proprotein convertase subtilisin kexin 9 phosphatidylinositol 3-kinase promyelocytic leukemia RNA interference |
| Raf1 S2P SARS-CoV | rapidly accelerated kinase 1 site-2 protease severe acute respiratory syndrome coronavirus |
| SCAP SKI-1/S1P SNARE | SREBP-cleavage activating protein subtilisin kexin isozyme-1/site-1 protease SNAP receptor |
| SRE | sterol regulatory element |
| SREBP STING | sterol regulatory element-binding protein stimulator of interferon genes |
| SVR S2P TAG TRIM16 UTR VLDL ZIKV 3D | sustained virologic response site-2 protease Triacylglycerol tri-partite-containing motif-16 untranslated region very low density lipoprotein Zika virus Three dimensional |
References
- Tauchi-Sato, K.; Ozeki, S.; Houjou, T.; Taguchi, R.; Fujimoto, T. The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J. Biol. Chem. 2002, 277, 44507–44512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweytick, D.; Athenstaedt, K.; Daum, G. Intracellular lipid particles of eukaryotic cells. Biochim. Biophys. Acta-Rev. Biomembr. 2000, 1469, 101–120. [Google Scholar] [CrossRef]
- Romanauska, A.; Köhler, A. The inner nuclear membrane is a metabolically active territory that generates nuclear lipid droplets. Cell 2018, 174, 700–715.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariri, H.; Rogers, S.; Ugrankar, R.; Liu, Y.L.; Feathers, J.R.; Henne, W.M. Lipid droplet biogenesis is spatially coordinated at ER–vacuole contacts under nutritional stress. EMBO Rep. 2018, 19, 57–72. [Google Scholar] [CrossRef]
- Wu, Y.W.; Mettling, C.; Wu, S.R.; Yu, C.Y.; Perng, G.C.; Lin, Y.S.; Lin, Y.L. Autophagy-associated dengue vesicles promote viral transmission avoiding antibody neutralization. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Welte, M.A. Expanding roles for lipid droplets. Curr. Biol. 2015, 25, R470–R481. [Google Scholar] [CrossRef] [Green Version]
- Roingeard, P.; Melo, R.C.N. Lipid droplet hijacking by intracellular pathogens. Cell. Microbiol. 2017, 19, e12688. [Google Scholar] [CrossRef]
- Hofmann, S.; Krajewski, M.; Scherer, C.; Scholz, V.; Mordhorst, V.; Truschow, P.; Schöbel, A.; Reimer, R.; Schwudke, D.; Herker, E. Complex lipid metabolic remodeling is required for efficient hepatitis C virus replication. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2018, 1863, 1041–1056. [Google Scholar] [CrossRef]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses exploit the lipid droplet protein AUP1 to trigger lipophagy and drive virus production. Cell Host Microbe 2018, 23, 819–831.e5. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, J.T.; Foung, S.; Muerhoff, A.S.; Bukh, J.; Simmonds, P. The GB viruses: A review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus pegivirus within the family flaviviridae. J. Gen. Virol. 2011, 92, 233–246. [Google Scholar] [CrossRef]
- Smith, D.B.; Meyers, G.; Bukh, J.; Gould, E.A.; Monath, T.; Muerhoff, A.S.; Pletnev, A.; Rico-Hesse, R.; Stapleton, J.T.; Simmonds, P.; et al. Proposed revision to the taxonomy of the genus pestivirus, family flaviviridae. J. Gen. Virol. 2017, 98, 2106–2112. [Google Scholar] [CrossRef]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef] [Green Version]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assunção-Miranda, I.; Barbosa-Lima, G.; Da Poian, A.T.; Bozza, P.T.; Gamarnik, A.V. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef]
- Shang, Z.; Song, H.; Shi, Y.; Qi, J.; Gao, G.F. Crystal structure of the capsid protein from zika virus. J. Mol. Biol. 2018, 430, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Filipe, A.; McLauchlan, J. Hepatitis C virus and lipid droplets: Finding a niche. Trends Mol. Med. 2015, 21, 34–42. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Cosset, F.-L.; Lohmann, V. Hepatitis C virus replication cycle. J. Hepatol. 2010, 53, 583–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.; Bartenschlager, R. Flaviviridae replication organelles: Oh, what a tangled web we weave. Annu. Rev. Virol. 2015, 2, 289–310. [Google Scholar] [CrossRef]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C virus RNA replication and assembly: Living on the fat of the land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Cortese, M.; Haselmann, U.; Tabata, K.; Romero-Brey, I.; Funaya, C.; Schieber, N.L.; Qiang, Y.; Bartenschlager, M.; Kallis, S.; et al. Spatiotemporal coupling of the hepatitis C virus replication cycle by creating a lipid droplet-proximal membranous replication compartment. Cell Rep. 2019, 27, 3602–3617.e5. [Google Scholar] [CrossRef] [Green Version]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; André, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Wrensch, F.; Crouchet, E.; Ligat, G.; Zeisel, M.B.; Keck, Z.Y.; Foung, S.K.H.; Schuster, C.; Baumert, T.F. Hepatitis C virus (HCV)-apolipoprotein interactions and immune evasion and their impact on HCV vaccine design. Front. Immunol. 2018, 9, 1436. [Google Scholar] [CrossRef] [PubMed]
- Vieyres, G.; Pietschmann, T. HCV pit stop at the lipid droplet: Refuel lipids and put on a lipoprotein coat before exit. Cells 2019, 8, 233. [Google Scholar] [CrossRef] [Green Version]
- Ishida, K.; Goto, S.; Ishimura, M.; Amanuma, M.; Hara, Y.; Suzuki, R.; Katoh, K.; Morita, E. Functional correlation between subcellular localizations of Japanese encephalitis virus capsid protein and virus production. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Gouilly, J.; Ferrat, Y.J.; Espino, A.; Glaziou, Q.; Cartron, G.; El Costa, H.; Al-Daccak, R.; Jabrane-Ferrat, N. Metabolic reprogramming by Zika virus provokes inflammation in human placenta. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Isken, O.; Langerwisch, U.; Schonherr, R.; Lamp, B.; Schroder, K.; Duden, R.; Rumenapf, T.H.; Tautz, N. Functional characterization of bovine viral diarrhea virus nonstructural protein 5A by reverse genetic analysis and live cell imaging. J. Virol. 2014, 88, 82–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lan, Y.; Sanyal, S. Modulation of lipid droplet metabolism-A potential target for therapeutic intervention in flaviviridae infections. Front. Microbiol. 2017, 8, 2286. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Olmstead, A.D.; Knecht, W.; Lazarov, I.; Dixit, S.B.; Jean, F. Human subtilase SKI-1/S1P is a master regulator of the HCV lifecycle and a potential host cell target for developing indirect-acting antiviral agents. PLoS Pathog. 2012, 8, e1002468. [Google Scholar] [CrossRef] [Green Version]
- Hyrina, A.; Meng, F.; McArthur, S.J.; Eivemark, S.; Nabi, I.R.; Jean, F. Human subtilisin kexin isozyme-1 (SKI-1)/Site-1 Protease (S1P) regulates cytoplasmic lipid droplet abundance: A potential target for indirect-acting anti-dengue virus agents. PLoS ONE 2017, 12, e0174483. [Google Scholar] [CrossRef]
- Hyrina, A.; Olmstead, A.D.; Steven, P.; Krajden, M.; Tam, E.; Jean, F. Treatment-induced viral cure of hepatitis C virus-infected patients involves a dynamic interplay among three important molecular players in lipid homeostasis: Circulating microRNA (miR)-24, miR-223, and proprotein convertase subtilisin/kexin Type 9. EBioMedicine 2017, 23, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawson, R.B.; Zelenski, N.G.; Nijhawan, D.; Ye, J.; Sakai, J.; Hasan, M.T.; Chang, T.Y.; Brown, M.S.; Goldstein, J.L. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell 1997, 1, 47–57. [Google Scholar] [CrossRef]
- Espenshade, P.J.; Cheng, D.; Goldstein, J.L.; Brown, M.S. Autocatalytic processing of site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 1999, 274, 22795–22804. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Lagace, T.A. PCSK9 and LDLR degradation: Regulatory mechanisms in circulation and in cells. Curr. Opin. Lipidol. 2014, 25, 387–393. [Google Scholar] [CrossRef]
- Shrivastava, S.; Steele, R.; Ray, R.; Ray, R.B. MicroRNAs: Role in hepatitis C virus pathogenesis. Genes Dis. 2015, 2, 35–45. [Google Scholar] [CrossRef] [Green Version]
- van der Grein, S.G.; Nolte-’t Hoen, E.N.M. “Small Talk” in the innate immune system via RNA-containing extracellular vesicles. Front. Immunol. 2014, 5, 542. [Google Scholar] [CrossRef] [Green Version]
- Ru, P.; Hu, P.; Geng, F.; Mo, X.; Cheng, C.; Yoo, J.Y.; Cheng, X.; Wu, X.; Guo, J.Y.; Nakano, I.; et al. Feedback loop regulation of SCAP/SREBP-1 by miR-29 modulates EGFR signaling-driven glioblastoma growth. Cell Rep. 2016, 16, 1527–1535. [Google Scholar] [CrossRef] [Green Version]
- Horie, T.; Nishino, T.; Baba, O.; Kuwabara, Y.; Nakao, T.; Nishiga, M.; Usami, S.; Izuhara, M.; Sowa, N.; Yahagi, N.; et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat. Commun. 2013, 4, 2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, R.; Wu, H.; Xiao, H.; Chen, X.; Willenbring, H.; Steer, C.J.; Song, G. Inhibition of microRNA-24 expression in liver prevents hepatic lipid accumulation and hyperlipidemia. Hepatology 2014, 60, 554–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michell, D.L.; Vickers, K.C. Lipoprotein carriers of microRNAs. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2016, 1861, 2069–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C Virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Liu, Q.; Sun, F.; Qiao, L. Hepatitis C virus nonstructural protein 5A perturbs lipid metabolism by modulating AMPK/SREBP-1c signaling. Lipids Health Dis. 2019, 18, 191. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Jun, H.J.; Wakita, T.; Cheong, J.H.; Hwang, S.B. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 2009, 284, 9237–9246. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Qiao, L.; Zhou, Y.; Babiuk, L.A.; Liu, Q. Hepatitis C virus nonstructural protein-5A activates sterol regulatory element-binding protein-1c through transcription factor Sp1. Biochem. Biophys. Res. Commun. 2010, 402, 549–553. [Google Scholar] [CrossRef]
- Fukasawa, M.; Tanaka, Y.; Sato, S.; Ono, Y.; Nitahara-Kasahara, Y.; Suzuki, T.; Miyamura, T.; Hanada, K.; Nishijima, M. Enhancement of de novo fatty acid biosynthesis in hepatic cell line Huh7 expressing hepatitis C virus core protein. Biol. Pharm. Bull. 2006, 29, 1958–1961. [Google Scholar] [CrossRef] [Green Version]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Pène, V.; Krishnamurthy, S.; Cha, H.; Liang, T.J. Hepatitis C virus infection activates an innate pathway involving IKK-α in lipogenesis and viral assembly. Nat. Med. 2013, 19, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Pène, V.; Li, Q.; Sodroski, C.; Hsu, C.-S.; Liang, T.J. Dynamic interaction of stress granules, DDX3X, and IKK-α mediates multiple functions in hepatitis C virus infection. J. Virol. 2015, 89, 5462–5477. [Google Scholar] [CrossRef] [Green Version]
- Vickers, K.C.; Landstreet, S.R.; Levin, M.G.; Shoucri, B.M.; Toth, C.L.; Taylor, R.C.; Palmisano, B.T.; Tabet, F.; Cui, H.L.; Rye, K.A.; et al. MicroRNA-223 coordinates cholesterol homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, 14518–14523. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, L.; Sun, J.; Chen, W.; Li, S.; Wang, Q.; Yu, H.; Xia, Z.; Jin, X.; Wang, C. Endoplasmic reticulum protein SCAP inhibits dengue virus NS2B3 protease by suppressing its K27-Linked polyubiquitylation. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, M.; Sureau, C.; Guévin, C.; Seidah, N.G.; Labonté, P. SKI-1/S1P inhibitor PF-429242 impairs the onset of HCV infection. Antiviral Res. 2015, 115, 94–104. [Google Scholar] [CrossRef]
- Corey, K.E.; Kane, E.; Munroe, C.; Barlow, L.L.; Zheng, H.; Chung, R.T. Hepatitis C virus infection and its clearance alter circulating lipids: Implications for long-term follow-up. Hepatology 2009, 50, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Albecka, A.; Belouzard, S.; de Beeck, A.O.; Descamps, V.; Goueslain, L.; Bertrand-Michel, J.; Tercé, F.; Duverlie, G.; Rouillé, Y.; Dubuisson, J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 2012, 55, 998–1007. [Google Scholar] [CrossRef]
- Labonté, P.; Begley, S.; Gúevin, C.; Asselin, M.C.; Nassoury, N.; Mayer, G.; Prat, A.; Seidah, N.G. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology 2009, 50, 17–24. [Google Scholar] [CrossRef]
- Seidah, N.G. New developments in proprotein convertase subtilisin-kexin 9’s biology and clinical implications. Curr. Opin. Lipidol. 2016, 27, 274–281. [Google Scholar] [CrossRef]
- Ingenito, F.; Roscigno, G.; Affnito, A.; Nuzzo, S.; Scognamiglio, I.; Quintavalle, C.; Condorelli, G. The role of Exo-miRNAs in cancer: A focus on therapeutic and diagnostic applications. Int. J. Mol. Sci. 2019, 20, 4687. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yu, F.; Ding, H.; Wang, Y.; Li, P.; Wang, K. Emerging function and clinical values of exosomal MicroRNAs in cancer. Mol. Ther. Nucleic Acids 2019, 16, 791–804. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Chu, H.; Chan, J.F.W.; Ye, Z.W.; Wen, L.; Yan, B.; Lai, P.M.; Tee, K.M.; Huang, J.; Chen, D.; et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Chung, J.; Wu, X.; Lambert, T.J.; Lai, Z.W.; Walther, T.C.; Farese, R.V. LDAF1 and Seipin form a lipid droplet assembly complex. Dev. Cell 2019, 51, 551–563.e7. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Layerenza, J.P.; González, P.; García De Bravo, M.M.; Polo, M.P.; Sisti, M.S.; Ves-Losada, A. Nuclear lipid droplets: A novel nuclear domain. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2013, 1831, 327–340. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Kawai, T.; Yoshikawa, Y.; Cheng, J.; Jokitalo, E.; Fujimoto, T. PML isoform II plays a critical role in nuclear lipid droplet formation. J. Cell Biol. 2016, 212, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef]
- Netsawang, J.; Noisakran, S.; Puttikhunt, C.; Kasinrerk, W.; Wongwiwat, W.; Malasit, P.; Yenchitsomanus, P.-T.; Limjindaporn, T. Nuclear localization of dengue virus capsid protein is required for DAXX interaction and apoptosis. Virus Res. 2010, 147, 275–283. [Google Scholar] [CrossRef]
- Falcón, V.; Acosta-Rivero, N.; Shibayama, M.; Chinea, G.; Gavilondo, J.V.; De La Rosa, M.C.; Menéndez, I.; Gra, B.; Dueñas-Carrera, S.; Viña, A.; et al. HCV core protein localizes in the nuclei of nonparenchymal liver cells from chronically HCV-infected patients. Biochem. Biophys. Res. Commun. 2005, 329, 1320–1328. [Google Scholar] [CrossRef]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C Virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef] [Green Version]
- Copley, S.D. An evolutionary perspective on protein moonlighting. Biochem. Soc. Trans. 2014, 42, 1684–1691. [Google Scholar] [CrossRef] [Green Version]
- Herzer, K.; Weyer, S.; Krammer, P.H.; Galle, P.R.; Hofmann, T.G. Hepatitis C virus core protein inhibits tumor suppressor protein promyelocytic leukemia function in human hepatoma cells. Cancer Res. 2005, 65, 10830–10837. [Google Scholar] [CrossRef] [Green Version]
- Herzer, K.; Carbow, A.; Sydor, S.; Sowa, J.-P.; Biesterfeld, S.; Hofmann, T.-G.; Galle, P.-R.; Gerken, G.; Canbay, A. Deficiency of the promyelocytic leukemia protein fosters Hepatitis C-associated hepatocarcinogenesis in mice. PLoS ONE 2012, 7, e44474. [Google Scholar] [CrossRef]
- Tanji, Y.; Kaneko, T.; Satoh, S.; Shimotohno, K. Phosphorylation of hepatitis C virus-encoded nonstructural protein NS5A. J. Virol. 1995, 69, 3980–3986. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Staschke, K.; De Francesco, R.; Tan, S.L. Phosphorylation of hepatitis C virus NS5A nonstructural protein: A new paradigm for phosphorylation-dependent viral RNA replication? Virology 2007, 364, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.-L.; Sheu, M.-L.; Yen, S.-H. Hepatitis C virus NS5A as a potential viral Bcl-2 homologue interacts with Bax and inhibits apoptosis in hepatocellular carcinoma. Int. J. Cancer 2003, 107, 65–73. [Google Scholar] [CrossRef]
- Zhang, Q.; Gong, R.; Qu, J.; Zhou, Y.; Liu, W.; Chen, M.; Liu, Y.; Zhu, Y.; Wu, J. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C Virus replication via attenuation of the interferon-JAK-STAT pathway. J. Virol. 2012, 86, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Sauter, D.; Himmelsbach, K.; Kriegs, M.; Carvajal Yepes, M.; Hildt, E. Localization determines function: N-terminally truncated NS5A fragments accumulate in the nucleus and impair HCV replication. J. Hepatol. 2009, 50, 861–871. [Google Scholar] [CrossRef]
- Zhao, Z.; Tao, M.; Han, W.; Fan, Z.; Imran, M.; Cao, S.; Ye, J. Nuclear localization of Zika virus NS5 contributes to suppression of type I interferon production and response. J. Gen. Virol. 2019. [Google Scholar] [CrossRef]
- Ng, I.H.W.; Chan, K.W.K.; Tan, M.J.A.; Gwee, C.P.; Smith, K.M.; Jeffress, S.J.; Saw, W.G.; Swarbrick, C.M.D.; Watanabe, S.; Jans, D.A.; et al. Zika Virus NS5 forms supramolecular nuclear bodies that sequester Importin-α and modulate the host immune and Pro-inflammatory response in neuronal cells. ACS Infect. Dis. 2019, 5, 932–948. [Google Scholar] [CrossRef]
- Chan, S.T.; Ou, J.H.J. Hepatitis C virus-induced autophagy and host innate immune response. Viruses 2017, 9, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, E.J.; Vevea, J.D.; Pon, L.A. Lipid droplet autophagy during energy mobilization, lipid homeostasis and protein quality control. Front. Biosci. Landmark 2018, 23, 1552–1563. [Google Scholar] [CrossRef] [Green Version]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Øverbye, A.; Brech, A.; Torgersen, M.L.; Jakobsen, I.S.; Sandvig, K.; Llorente, A. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell. Mol. Life Sci. 2016, 73, 4717–4737. [Google Scholar] [CrossRef]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef]
- Klemm, E.J.; Spooner, E.; Ploegh, H.L. Dual role of Ancient Ubiquitous Protein 1 (AUP1) in lipid droplet accumulation and Endoplasmic Reticulum (ER) protein quality control. J. Biol. Chem. 2011, 286, 37602–37614. [Google Scholar] [CrossRef] [Green Version]
- Lennemann, N.J.; Coyne, C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 2017, 13, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.W.; Li, Z.L.; Yuan, S. The role of secretory autophagy in Zika virus transfer through the placental barrier. Front. Cell. Infect. Microbiol. 2017, 6, 206. [Google Scholar] [CrossRef]
- Shen, J.; Huang, C.K.; Yu, H.; Shen, B.; Zhang, Y.; Liang, Y.; Li, Z.; Feng, X.; Zhao, J.; Duan, L.; et al. The role of exosomes in hepatitis, liver cirrhosis and hepatocellular carcinoma. J. Cell. Mol. Med. 2017, 21, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from Hepatitis C Infected Patients Transmit HCV Infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.-C.; Ray, R.; Ray, R.B. Knockdown of autophagy inhibits infectious Hepatitis C virus release by the exosomal pathway. J. Virol. 2016, 90, 1387–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, K.; Liu, Y.; Xu, Y.; Zhang, F.; Yang, H.; Liu, J.; Pan, T.; Chen, J.; Wu, M.; et al. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat. Immunol. 2013, 14, 793–803. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, P. The lipid droplet: A conserved cellular organelle. Protein Cell 2017, 8, 796–800. [Google Scholar] [CrossRef]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 225-IN6. [Google Scholar] [CrossRef]
- Melvin, S.L.; Dawson, G.J.; Carrick, R.J.; Schlauder, G.G.; Heynen, C.A.; Mushahwar, I.K. Biophysical characterization of GB virus C from human plasma. J. Virol. Methods 1998, 71, 147–157. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 1–14. [Google Scholar] [CrossRef]
- Silvas, J.A.; Jureka, A.S.; Nicolini, A.M.; Chvatal, S.A.; Basler, C.F. Inhibitors of VPS34 and lipid metabolism suppress SARS-CoV-2 replication 1. bioRxiv 2020. [Google Scholar] [CrossRef]
- Plummer, E.; Buck, M.D.; Sanchez, M.; Greenbaum, J.A.; Turner, J.; Grewal, R.; Klose, B.; Sampath, A.; Warfield, K.L.; Peters, B.; et al. Dengue virus evolution under a host-targeted antiviral. J. Virol. 2015, 89, 5592–5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, M.B.; Lupberger, J.; Fofana, I.; Baumert, T.F. Host-targeting agents for prevention and treatment of chronic hepatitis C-Perspectives and challenges. J. Hepatol. 2013, 58, 375–384. [Google Scholar] [CrossRef]
- Dubey, R.; Stivala, C.E.; Nguyen, H.Q.; Goo, Y.H.; Paul, A.; Carette, J.E.; Trost, B.M.; Rohatgi, R. Lipid droplets can promote drug accumulation and activation. Nat. Chem. Biol. 2020, 16, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Daemen, S.; Van Polanen, N.; Hesselink, M.K.C. The effect of diet and exercise on lipid droplet dynamics in human muscle tissue. J. Exp. Biol. 2018, 121, jeb167015. [Google Scholar] [CrossRef] [Green Version]
- Chiu, T.H.; Lin, M.-N.; Pan, W.-H.; Chen, Y.-C.; Lin, C.-L. Vegetarian diet, food substitution, and nonalcoholic fatty liver. Tzu-Chi Med. J. 2018, 30, 102–109. [Google Scholar] [CrossRef]
- Pérez-Martínez, P.; Mikhailidis, D.P.; Athyros, V.G.; Bullo, M.; Couture, P.; Covas, M.I.; de Koning, L.; Delgado-Lista, J.; Díaz-López, A.; Drevon, C.A.; et al. Lifestyle recommendations for the prevention and management of metabolic syndrome: An international panel recommendation. Nutr. Rev. 2017, 75, 307–326. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Virus Component | Hepatitis C Virus | Dengue and Zika Viruses |
|---|---|---|
| Genome | +ssRNA | +ssRNA |
| Capsid protein | Core * | C * |
| Envelope proteins | E1, E2 | prM, E |
| Nonstructural proteins | NS2, NS3, NS4A, NS4B Δ, NS5A *, NS5B | NS1, NS2A, NS2B Δ, NS3 Δ, NS4A Δ, 2k, NS4B Δ, NS5 |
| Virus | SREBP-Hijacking Action | Viral Factor (s) | References |
|---|---|---|---|
| HCV | Increases SREBP-1 and SREBP-2 expression in in vitro infection models | NS5A, NS4B, 3′ UTR | [45,46,47,50,51] |
| Activates SREBP signaling in in vitro infection models | Core, NS4B, NS5A | [44,46,47] | |
| Reduces PCSK9, miR-24 and miR-223 concentrations in non-responders to antiviral treatment | Unknown | [31,42,52] | |
| DENV | Hijacks host SKI-1/S1P to activate SREBP pathway | Unknown | [30] |
| Cleaves host protein STING to prevent expression of interferon genes | NS2B, NS3 | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cloherty, A.P.M.; Olmstead, A.D.; Ribeiro, C.M.S.; Jean, F. Hijacking of Lipid Droplets by Hepatitis C, Dengue and Zika Viruses—From Viral Protein Moonlighting to Extracellular Release. Int. J. Mol. Sci. 2020, 21, 7901. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217901
Cloherty APM, Olmstead AD, Ribeiro CMS, Jean F. Hijacking of Lipid Droplets by Hepatitis C, Dengue and Zika Viruses—From Viral Protein Moonlighting to Extracellular Release. International Journal of Molecular Sciences. 2020; 21(21):7901. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217901
Chicago/Turabian StyleCloherty, Alexandra P.M., Andrea D. Olmstead, Carla M.S. Ribeiro, and François Jean. 2020. "Hijacking of Lipid Droplets by Hepatitis C, Dengue and Zika Viruses—From Viral Protein Moonlighting to Extracellular Release" International Journal of Molecular Sciences 21, no. 21: 7901. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217901