
Hydroxygenkwanin Increases the Sensitivity of Liver Cancer Cells to Chemotherapy by Inhibiting DNA Damage Response in Mouse Xenograft Models

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
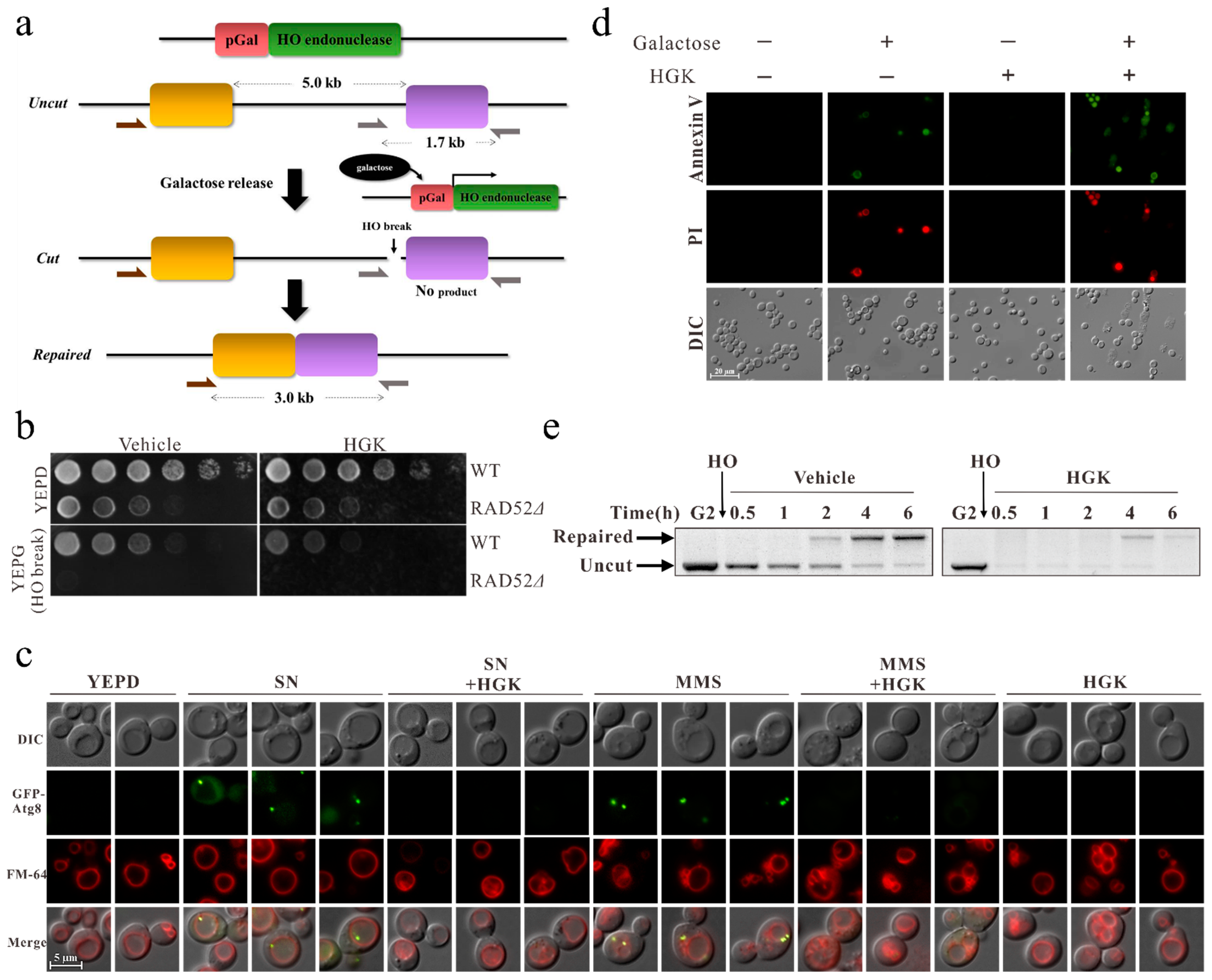
2.1. HGK Inhibits DDR and Promotes Apoptosis in Yeast Cells
2.2. HGK Inhibits DDR of Liver Cancer Cell Lines
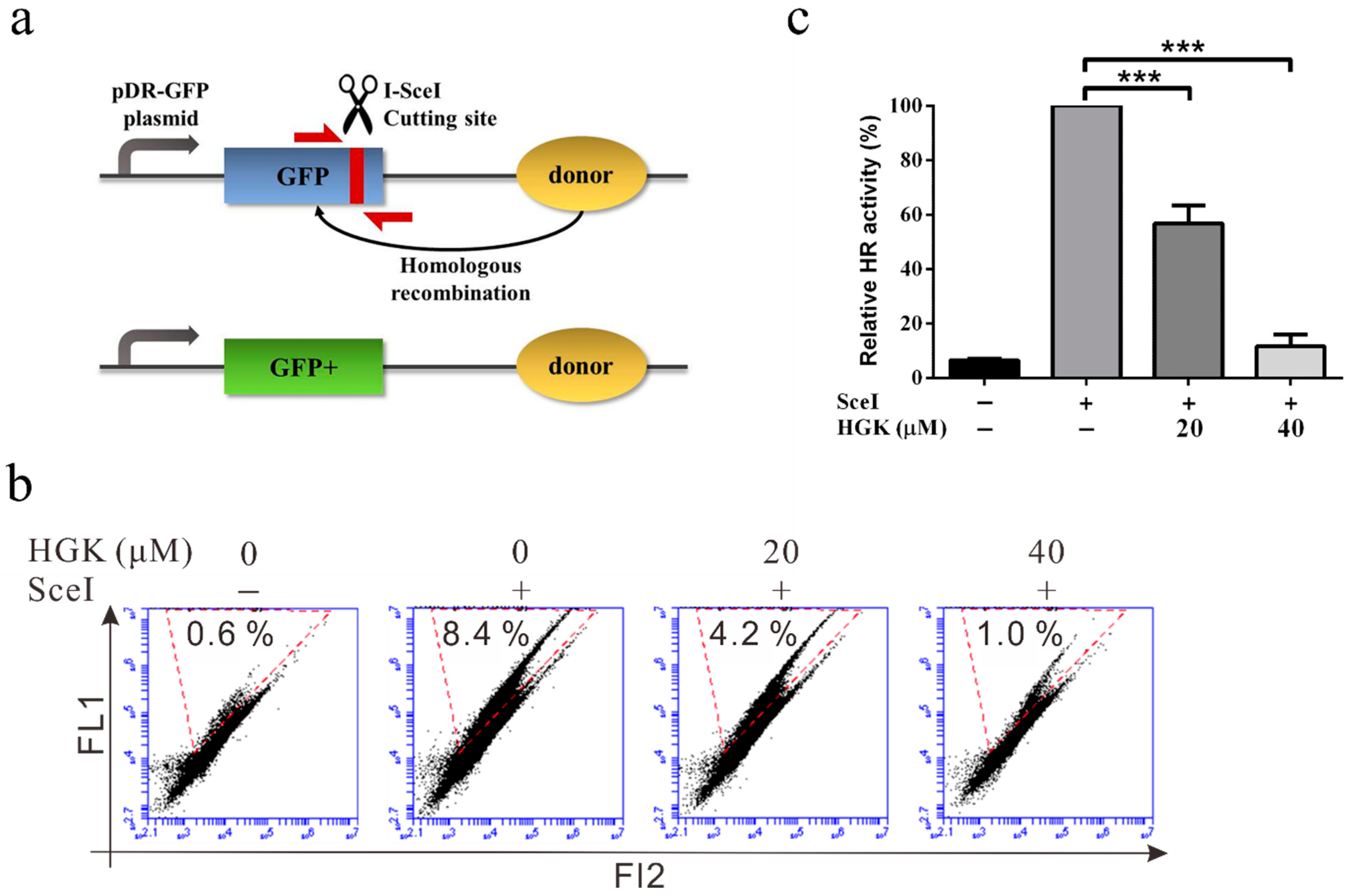
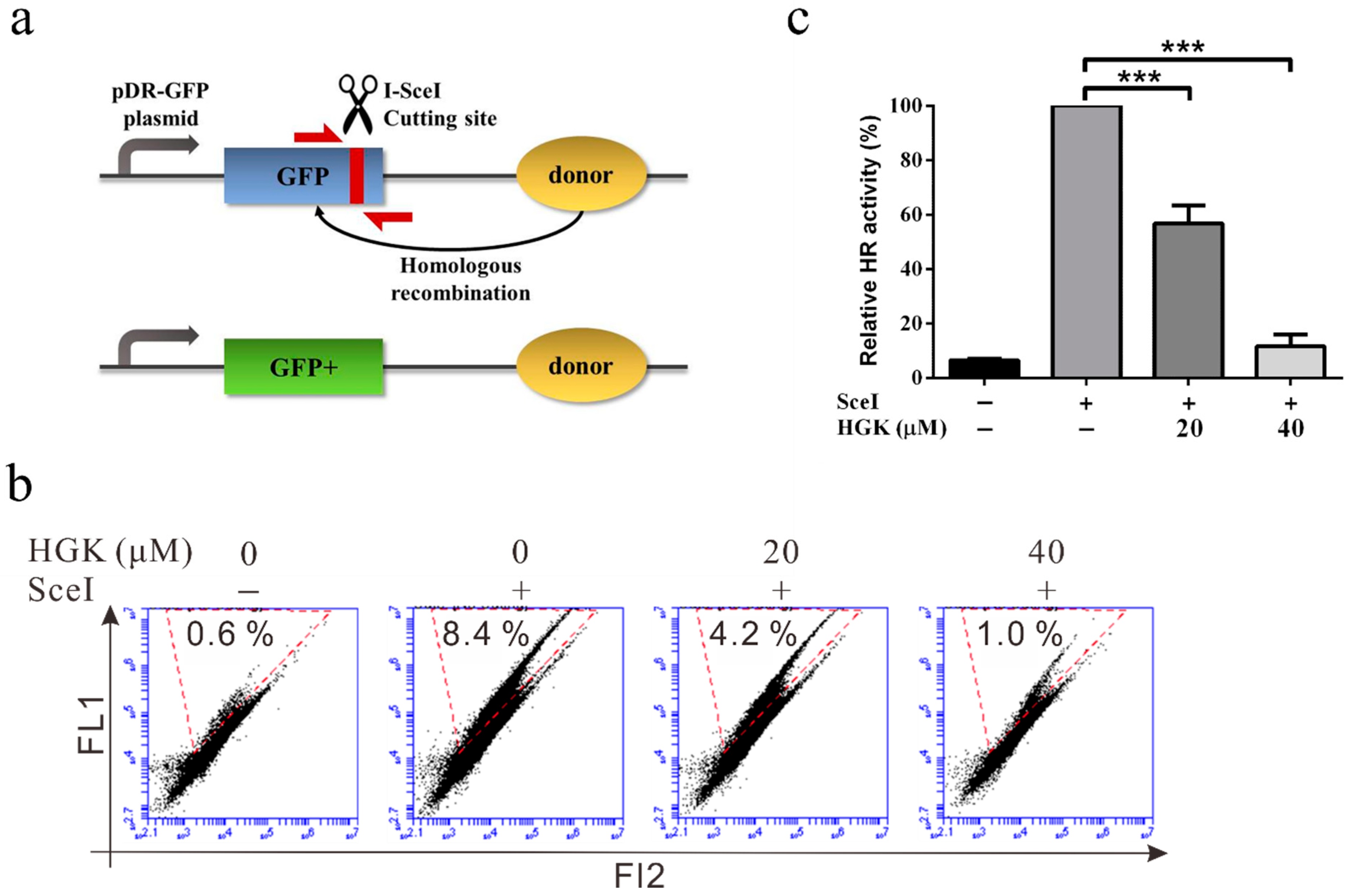
2.3. HGK Inhibits HR Repair in Liver Cancer Cell Lines
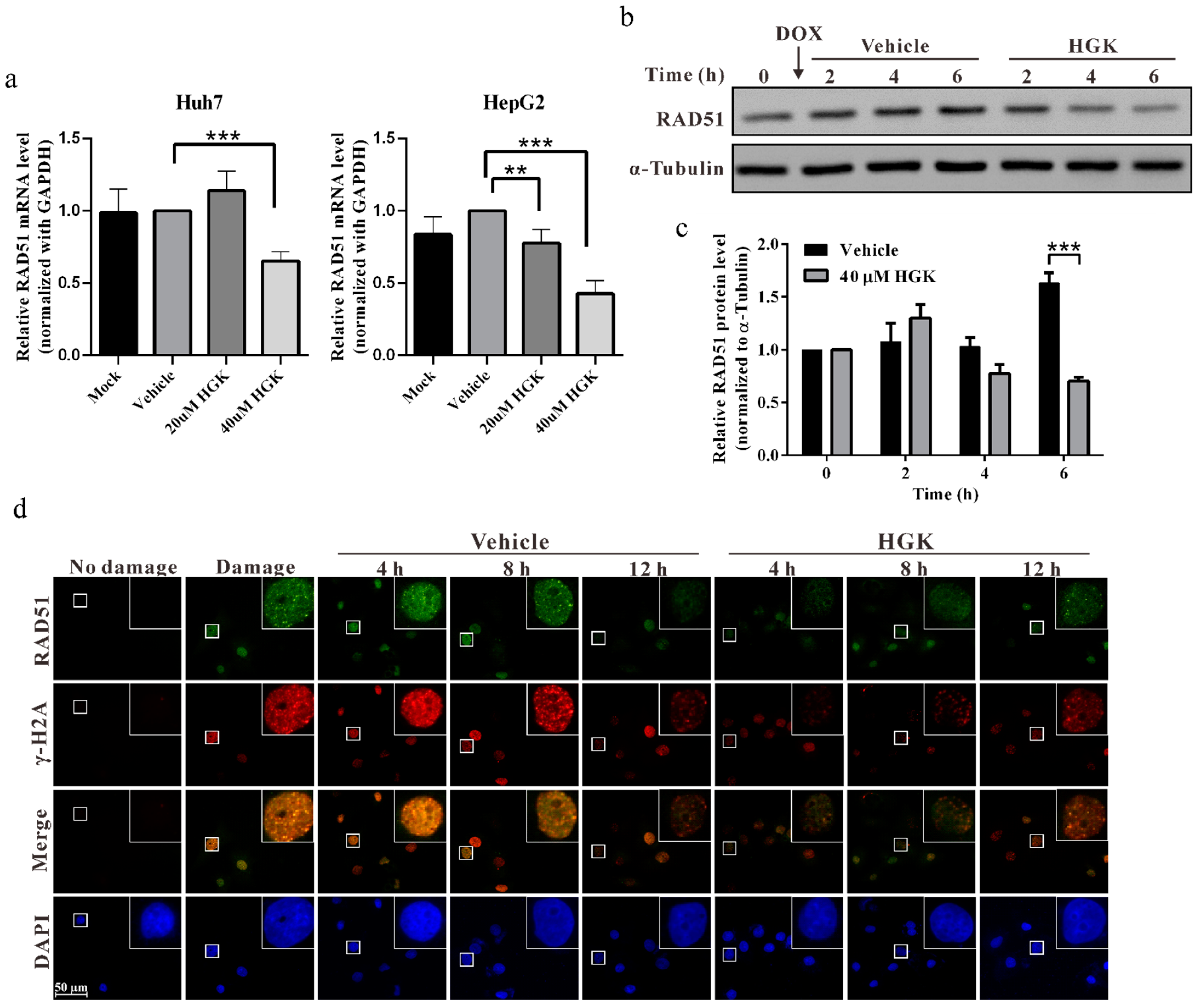
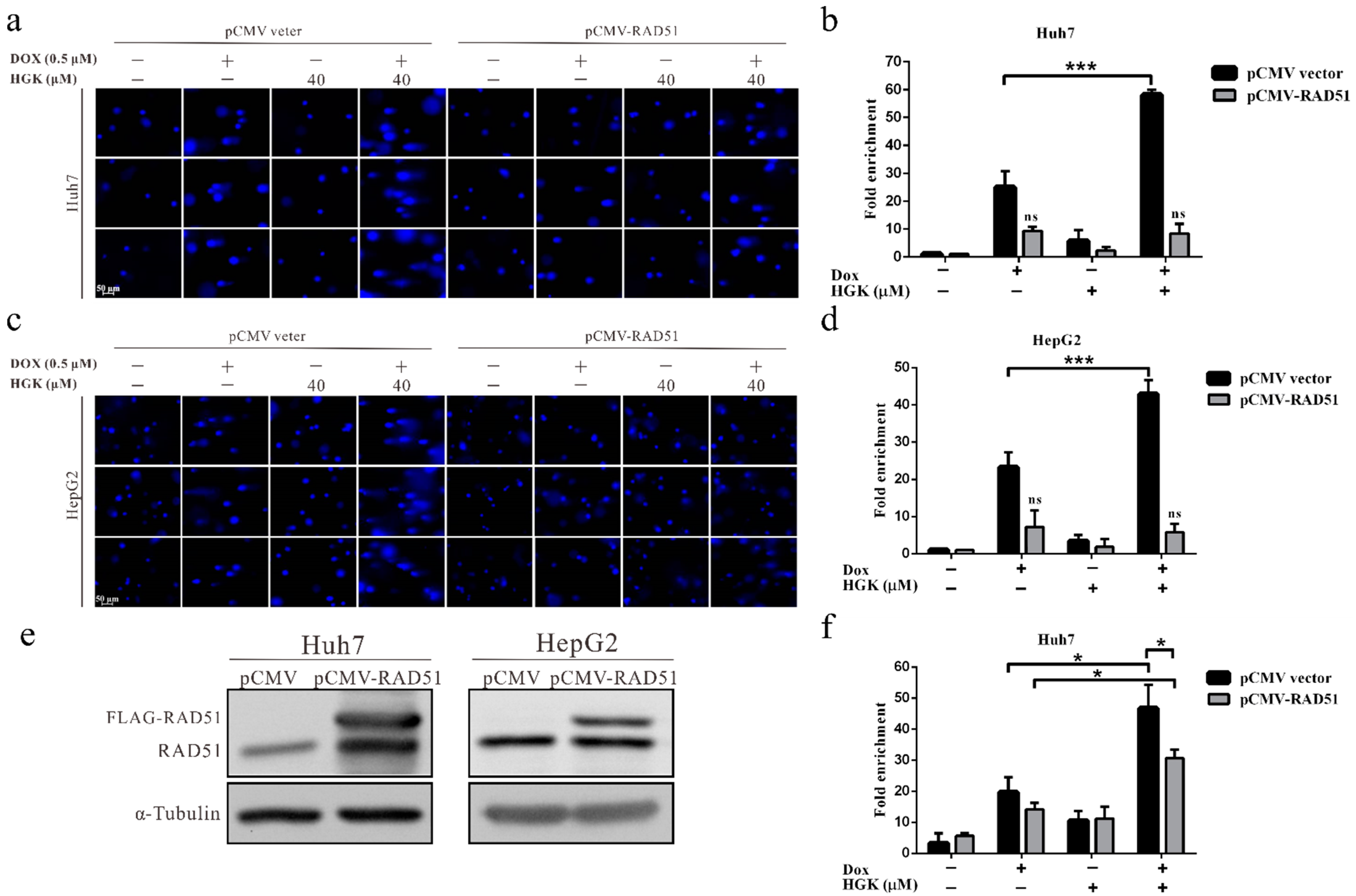
2.4. HGK Inhibits HR Progression by Suppressing the Expression of RAD51
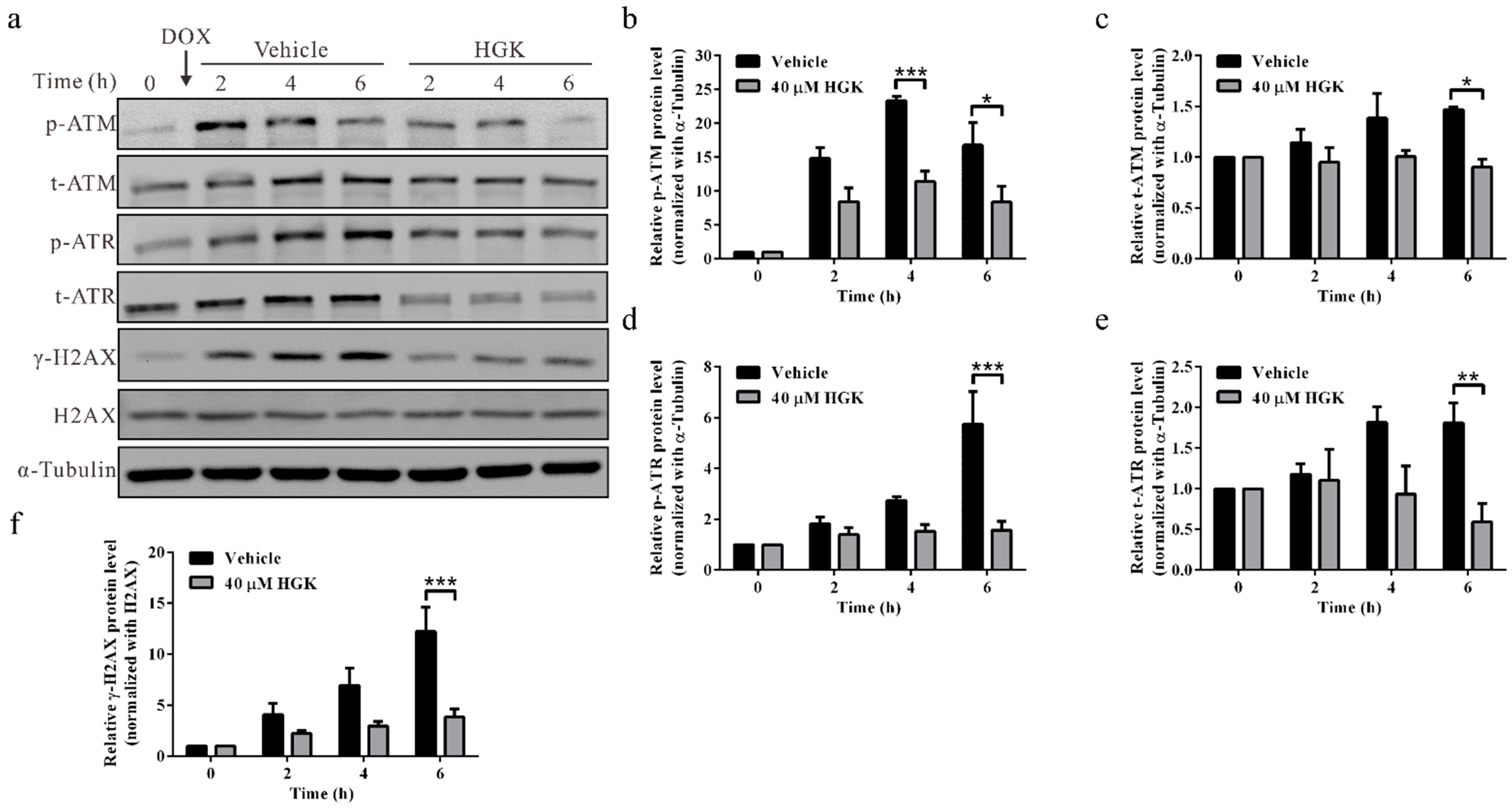
2.5. HGK Inhibits Phosphorylation of DNA Damage Checkpoint Proteins ATM/ATR
2.6. HGK Improves the Efficacy of Chemotherapy in HCC Cell Lines
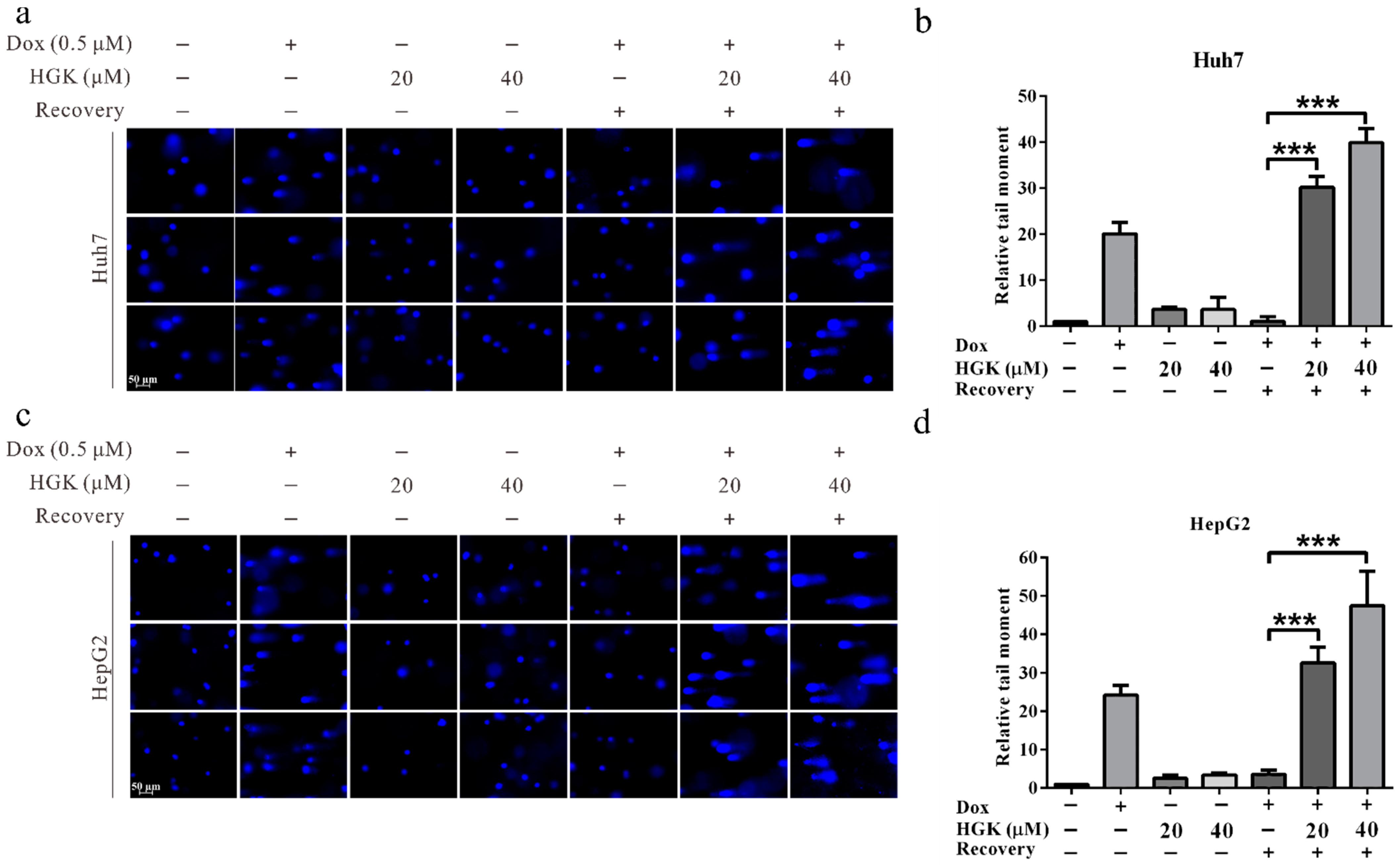
2.7. HGK Improves the Efficacy of Doxorubicin in HCC Cell Lines by Inhibiting RAD51-Mediated DDR
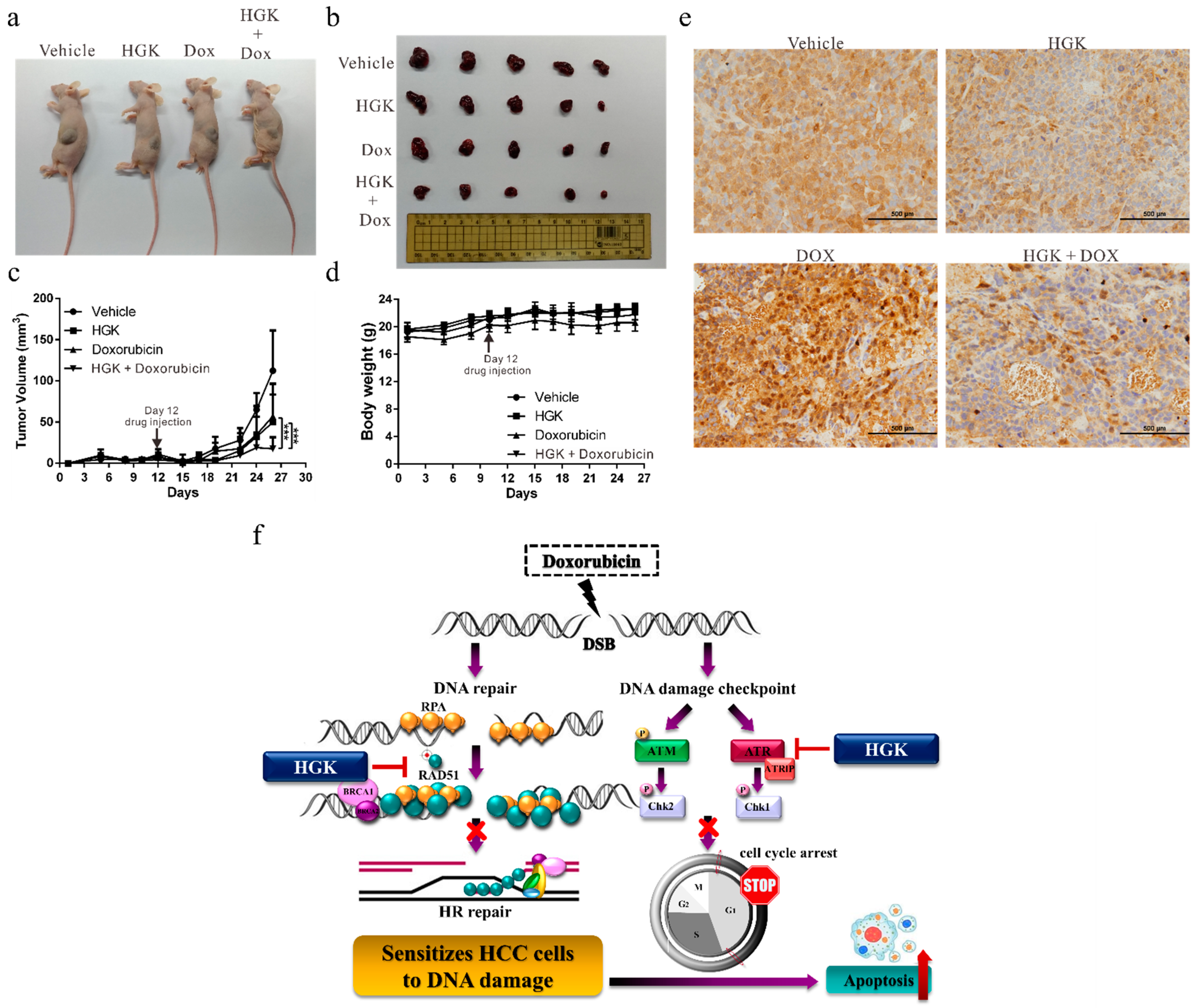
2.8. HGK Increases the Sensitivity of Liver Cancer Cells to Doxorubicin In Vivo
3. Discussion
4. Materials and Methods
4.1. Yeast Strains, Cell Lines, Antibodies, Plasmids, and Drugs
4.2. HO Induction
4.3. DNA Damage Sensitivity Plate Assay
4.4. DNA Cutting and Repair Analysis
4.5. Fluorescence Microscopy
4.6. Real-Time RT-PCR
4.7. Western Blotting Analysis
4.8. Cell Proliferation Assay
4.9. Cell Migration and Invasion Assays
4.10. Apoptosis Assay
4.11. Comet Assay
4.12. HR Assay
4.13. Cell-Cycle Analysis
4.14. Animal Experiments
4.15. Immunohistochemistry
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinrichsen, I.; Kemp, M.; Peveling-Oberhag, J.; Passmann, S.; Plotz, G.; Zeuzem, S.; Brieger, A. Promoter methylation of MLH1, PMS2, MSH2 and p16 is a phenomenon of advanced-stage HCCs. PLoS ONE 2014, 9, e84453. [Google Scholar] [CrossRef]
- Rao, C.V.; Asch, A.S.; Yamada, H.Y. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis 2017, 38, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Xiong, C.; Chen, Y.; Zhang, C.; Bai, D. Identification of Rad51 as a prognostic biomarker correlated with immune infiltration in hepatocellular carcinoma. Bioengineered 2021, 12, 2664–2675. [Google Scholar] [CrossRef]
- Anand, S.K.; Sharma, A.; Singh, N.; Kakkar, P. Entrenching role of cell cycle checkpoints and autophagy for maintenance of genomic integrity. DNA Repair 2020, 86, 102748. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Burdak-Rothkamm, S.; Rothkamm, K. Radiation-induced bystander and systemic effects serve as a unifying model system for genotoxic stress responses. Mutat. Res. 2018, 778, 13–22. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Kargapolova, Y.; Rehimi, R.; Kayserili, H.; Bruhl, J.; Sofiadis, K.; Zirkel, A.; Palikyras, S.; Mizi, A.; Li, Y.; Yigit, G.; et al. Overarching control of autophagy and DNA damage response by CHD6 revealed by modeling a rare human pathology. Nat. Commun. 2021, 12, 3014. [Google Scholar] [CrossRef] [PubMed]
- Chitikova, Z.V.; Gordeev, S.A.; Bykova, T.V.; Zubova, S.G.; Pospelov, V.A.; Pospelova, T.V. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces reversible senescence associated with mTOR downregulation and expression of stem cell markers. Cell Cycle 2014, 13, 1424–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef]
- Prokhorova, E.A.; Egorshina, A.Y.; Zhivotovsky, B.; Kopeina, G.S. The DNA-damage response and nuclear events as regulators of nonapoptotic forms of cell death. Oncogene 2020, 39, 1–16. [Google Scholar] [CrossRef]
- Lee, T.H.; Kang, T.H. DNA Oxidation and Excision Repair Pathways. Int. J. Mol. Sci. 2019, 20, 6092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza-Aghazadeh-Attari, M.; Recio, M.J.; Darband, S.G.; Kaviani, M.; Safa, A.; Mihanfar, A.; Sadighparvar, S.; Karimian, A.; Alemi, F.; Majidinia, M.; et al. DNA damage response and breast cancer development: Possible therapeutic applications of ATR, ATM, PARP, BRCA1 inhibition. DNA Repair 2021, 98, 103032. [Google Scholar] [CrossRef] [PubMed]
- Begam, N.; Jamil, K.; Raju, S.G. Promoter Hypermethylation of the ATM Gene as a Novel Biomarker for Breast Cancer. Asian Pac. J. Cancer Prev. 2017, 18, 3003–3009. [Google Scholar] [CrossRef]
- Podralska, M.; Ziolkowska-Suchanek, I.; Zurawek, M.; Dzikiewicz-Krawczyk, A.; Slomski, R.; Nowak, J.; Stembalska, A.; Pesz, K.; Mosor, M. Genetic variants in ATM, H2AFX and MRE11 genes and susceptibility to breast cancer in the polish population. BMC Cancer 2018, 18, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallajian, Z.; Mahjoubi, F.; Nafissi, N. Simultaneous ATM/BRCA1/RAD51 expression variations associated with prognostic factors in Iranian sporadic breast cancer patients. Breast Cancer 2017, 24, 624–634. [Google Scholar] [CrossRef]
- Lozano, R.; Castro, E.; Aragon, I.M.; Cendon, Y.; Cattrini, C.; Lopez-Casas, P.P.; Olmos, D. Genetic aberrations in DNA repair pathways: A cornerstone of precision oncology in prostate cancer. Br. J. Cancer 2021, 124, 552–563. [Google Scholar] [CrossRef]
- Lieberman, H.B.; Panigrahi, S.K.; Hopkins, K.M.; Wang, L.; Broustas, C.G. p53 and RAD9, the DNA Damage Response, and Regulation of Transcription Networks. Radiat. Res. 2017, 187, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Ding, Y.; Li, D.D.; Peng, R.Q.; Feng, G.K.; Zeng, Y.X.; Zhu, X.F.; Zhang, X.S. The overexpression of ERCC-1 is involved in the resistance of lung cancer cells to cetuximab combined with DDP. Cancer Biol. Ther. 2009, 8, 1914–1921. [Google Scholar] [CrossRef] [Green Version]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Abubakar, A.; Lim, S.H.; Ng, W.; Lee, M.; de Souza, P.; Shin, J.S.; et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) complex in rectal cancer correlates with poor response to neoadjuvant radiotherapy and prognosis. BMC Cancer 2018, 18, 869. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.B.; Chen, Y.; Meng, X.D.; Yu, P.; He, X.; Li, J. Nucleotide Excision Repair Factor XPC Ameliorates Prognosis by Increasing the Susceptibility of Human Colorectal Cancer to Chemotherapy and Ionizing Radiation. Front. Oncol. 2018, 8, 290. [Google Scholar] [CrossRef]
- Nagathihalli, N.S.; Nagaraju, G. RAD51 as a potential biomarker and therapeutic target for pancreatic cancer. Biochim. Biophys. Acta 2011, 1816, 209–218. [Google Scholar] [CrossRef]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef]
- Kopa, P.; Macieja, A.; Galita, G.; Witczak, Z.J.; Poplawski, T. DNA Double Strand Breaks Repair Inhibitors: Relevance as Potential New Anticancer Therapeutics. Curr. Med. Chem. 2019, 26, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Velic, D.; Charlier, C.; Popova, M.; Jaunet-Lahary, T.; Bouchouireb, Z.; Henry, S.; Weigel, P.; Masson, J.Y.; Laurent, A.; Nabiev, I.; et al. Interactions of the Rad51 inhibitor DIDS with human and bovine serum albumins: Optical spectroscopy and isothermal calorimetry approaches. Biochimie 2019, 167, 187–197. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Tang, L.; Tanaka, N.; Gleber-Netto, F.O.; Bartels, M.D.; Wang, L.; McGrail, D.J.; Lin, S.Y.; Frank, S.J.; et al. Combined Inhibition of Rad51 and Wee1 Enhances Cell Killing in HNSCC Through Induction of Apoptosis Associated with Excessive DNA Damage and Replication Stress. Mol. Cancer Ther. 2021, 20, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Aubry, A.; Pearson, J.D.; Huang, K.; Livne-Bar, I.; Ahmad, M.; Jagadeesan, M.; Khetan, V.; Ketela, T.; Brown, K.R.; Yu, T.; et al. Functional genomics identifies new synergistic therapies for retinoblastoma. Oncogene 2020, 39, 5338–5357. [Google Scholar] [CrossRef] [PubMed]
- Berte, N.; Piee-Staffa, A.; Piecha, N.; Wang, M.; Borgmann, K.; Kaina, B.; Nikolova, T. Targeting Homologous Recombination by Pharmacological Inhibitors Enhances the Killing Response of Glioblastoma Cells Treated with Alkylating Drugs. Mol. Cancer Ther. 2016, 15, 2665–2678. [Google Scholar] [CrossRef] [Green Version]
- Petsalaki, E.; Zachos, G. DNA damage response proteins regulating mitotic cell division: Double agents preserving genome stability. FEBS J. 2020, 287, 1700–1721. [Google Scholar] [CrossRef] [PubMed]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, A.; Kim, J.; Walker, C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010, 6, 672–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef]
- Teng, P.N.; Bateman, N.W.; Darcy, K.M.; Hamilton, C.A.; Maxwell, G.L.; Bakkenist, C.J.; Conrads, T.P. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol. Oncol. 2015, 136, 554–561. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, D.; Biswasroy, P.; Sahu, A.; Sahu, D.K.; Ghosh, G.; Rath, G. Recent advances in herbal nanomedicines for cancer treatment. Curr. Mol. Pharm. 2021, 14, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Gezici, S.; Sekeroglu, N. Current Perspectives in the Application of Medicinal Plants Against Cancer: Novel Therapeutic Agents. Anticancer Agents Med. Chem. 2019, 19, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.T.; Liu, X.D.; Zhan, Z.P.; Wu, Q.J. Sulforaphane enhances the cisplatin sensitivity through regulating DNA repair and accumulation of intracellular cisplatin in ovarian cancer cells. Exp. Cell Res. 2020, 393, 112061. [Google Scholar] [CrossRef] [PubMed]
- Leon-Galicia, I.; Diaz-Chavez, J.; Garcia-Villa, E.; Uribe-Figueroa, L.; Hidalgo-Miranda, A.; Herrera, L.A.; Alvarez-Rios, E.; Garcia-Mena, J.; Gariglio, P. Resveratrol induces downregulation of DNA repair genes in MCF-7 human breast cancer cells. Eur. J. Cancer Prev. 2013, 22, 11–20. [Google Scholar] [CrossRef]
- Ruiz, G.; Valencia-Gonzalez, H.A.; Leon-Galicia, I.; Garcia-Villa, E.; Garcia-Carranca, A.; Gariglio, P. Inhibition of RAD51 by siRNA and Resveratrol Sensitizes Cancer Stem Cells Derived from HeLa Cell Cultures to Apoptosis. Stem Cells Int. 2018, 2018, 2493869. [Google Scholar] [CrossRef]
- Kuo, C.H.; Leu, Y.L.; Wang, T.H.; Tseng, W.C.; Feng, C.H.; Wang, S.H.; Chen, C.C. A novel DNA repair inhibitor, diallyl disulfide (DADS), impairs DNA resection during DNA double-strand break repair by reducing Sae2 and Exo1 levels. DNA Repair 2019, 82, 102690. [Google Scholar] [CrossRef]
- Li, S.; Chou, G.; Hseu, Y.; Yang, H.; Kwan, H.; Yu, Z. Isolation of anticancer constituents from flos genkwa (Daphne genkwa Sieb.et Zucc.) through bioassay-guided procedures. Chem. Cent. J. 2013, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Zhang, D.D. Anti-inflammatory effects of 81 chinese herb extracts and their correlation with the characteristics of traditional chinese medicine. Evid. Based Complement. Altern. Med. 2014, 2014, 985176. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.S.; Yin, L.H.; Xu, L.N.; Peng, J.Y.; Zhou, H.; Kang, W. Synergistic anti-glioma effect of Hydroxygenkwanin and Apigenin in vitro. Chem. Biol. Interact. 2013, 206, 346–355. [Google Scholar] [CrossRef]
- Li, N.; Liu, J.H.; Zhang, J.; Yu, B.Y. Comparative evaluation of cytotoxicity and antioxidative activity of 20 flavonoids. J. Agric. Food Chem. 2008, 56, 3876–3883. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, C.C.; Chuang, W.Y.; Leu, Y.L.; Ueng, S.H.; Hsueh, C.; Yeh, C.T.; Wang, T.H. Hydroxygenkwanin Inhibits Class I HDAC Expression and Synergistically Enhances the Antitumor Activity of Sorafenib in Liver Cancer Cells. Front. Oncol. 2020, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Chou, L.F.; Chen, C.Y.; Yang, W.H.; Chen, C.C.; Chang, J.L.; Leu, Y.L.; Liou, M.J.; Wang, T.H. Suppression of Hepatocellular Carcinoma Progression through FOXM1 and EMT Inhibition via Hydroxygenkwanin-Induced miR-320a Expression. Biomolecules 2019, 10, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatkowska, M.; Krupa, R. Regulation of DNA Damage Response and Homologous Recombination Repair by microRNA in Human Cells Exposed to Ionizing Radiation. Cancers 2020, 12, 1838. [Google Scholar] [CrossRef]
- Laurini, E.; Marson, D.; Fermeglia, A.; Aulic, S.; Fermeglia, M.; Pricl, S. Role of Rad51 and DNA repair in cancer: A molecular perspective. Pharm. Ther. 2020, 208, 107492. [Google Scholar] [CrossRef]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol. Cell Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef] [Green Version]
- Orhan, E.; Velazquez, C.; Tabet, I.; Sardet, C.; Theillet, C. Regulation of RAD51 at the Transcriptional and Functional Levels: What Prospects for Cancer Therapy? Cancers 2021, 13, 2930. [Google Scholar] [CrossRef]
- Ward, A.; Khanna, K.K.; Wiegmans, A.P. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat. Rev. 2015, 41, 35–45. [Google Scholar] [CrossRef]
- Ko, J.C.; Chen, J.C.; Wang, T.J.; Zheng, H.Y.; Chen, W.C.; Chang, P.Y.; Lin, Y.W. Astaxanthin down-regulates Rad51 expression via inactivation of AKT kinase to enhance mitomycin C-induced cytotoxicity in human non-small cell lung cancer cells. Biochem. Pharm. 2016, 105, 91–100. [Google Scholar] [CrossRef]
- Li, Y.; Liu, F.; Wang, Y.; Li, D.; Guo, F.; Xu, L.; Zeng, Z.; Zhong, X.; Qian, K. Rapamycin-induced autophagy sensitizes A549 cells to radiation associated with DNA damage repair inhibition. Thorac. Cancer 2016, 7, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Yuk, J.M.; Yoshimori, T.; Jo, E.K. Autophagy and bacterial infectious diseases. Exp. Mol. Med. 2012, 44, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef] [PubMed]
- Pena-Oyarzun, D.; Reyes, M.; Hernandez-Caceres, M.P.; Kretschmar, C.; Morselli, E.; Ramirez-Sarmiento, C.A.; Lavandero, S.; Torres, V.A.; Criollo, A. Role of Autophagy in the Microenvironment of Oral Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 602661. [Google Scholar] [CrossRef]
- Lin, T.; Zhang, Q.; Yuan, A.; Wang, B.; Zhang, F.; Ding, Y.; Cao, W.; Chen, W.; Guo, H. Synergy of Tumor Microenvironment Remodeling and Autophagy Inhibition to Sensitize Radiation for Bladder Cancer Treatment. Theranostics 2020, 10, 7683–7696. [Google Scholar] [CrossRef]
- Vempati, R.K.; Malla, R.R. Autophagy-Induced Drug Resistance in Liver Cancer. Crit. Rev. Oncog. 2020, 25, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Chen, C.Y.; Ueng, S.H.; Hsueh, C.; Yeh, C.T.; Ho, J.Y.; Chou, L.F.; Wang, T.H. Corylin increases the sensitivity of hepatocellular carcinoma cells to chemotherapy through long noncoding RNA RAD51-AS1-mediated inhibition of DNA repair. Cell Death Dis. 2018, 9, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genotype | Source |
|---|---|---|
| YMV045 | ho hml∆::ADE1 mata∆::hisG hmr∆::ade1 leu2::leu2 (Asp-718-SalI)-URA3-pBR322-HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG) | James Haber |
| YMV046 | ho hml∆::ADE1 mata∆::hisG hmr∆::ADE1 leu2:HOcs ade3::GAL::HO ade1 lys5 ura3-52 trp1 (trp1::hisG) rad52∆::HPH (hygro) | James Haber |
| RLY004 | BY4741-atg8 transform with PRS416 GFP-Atg8 in URA drop media | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-C.; Chen, C.-Y.; Cheng, S.-F.; Shieh, T.-M.; Leu, Y.-L.; Chuang, W.-Y.; Liu, K.-T.; Ueng, S.-H.; Shih, Y.-H.; Chou, L.-F.; et al. Hydroxygenkwanin Increases the Sensitivity of Liver Cancer Cells to Chemotherapy by Inhibiting DNA Damage Response in Mouse Xenograft Models. Int. J. Mol. Sci. 2021, 22, 9766. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189766
Chen C-C, Chen C-Y, Cheng S-F, Shieh T-M, Leu Y-L, Chuang W-Y, Liu K-T, Ueng S-H, Shih Y-H, Chou L-F, et al. Hydroxygenkwanin Increases the Sensitivity of Liver Cancer Cells to Chemotherapy by Inhibiting DNA Damage Response in Mouse Xenograft Models. International Journal of Molecular Sciences. 2021; 22(18):9766. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189766
Chicago/Turabian StyleChen, Chin-Chuan, Chi-Yuan Chen, Shu-Fang Cheng, Tzong-Ming Shieh, Yann-Lii Leu, Wen-Yu Chuang, Kuang-Ting Liu, Shir-Hwa Ueng, Yin-Hwa Shih, Li-Fang Chou, and et al. 2021. "Hydroxygenkwanin Increases the Sensitivity of Liver Cancer Cells to Chemotherapy by Inhibiting DNA Damage Response in Mouse Xenograft Models" International Journal of Molecular Sciences 22, no. 18: 9766. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189766