
Investigating Pathogenetic Mechanisms of Alzheimer’s Disease by Systems Biology Approaches for Drug Discovery


Abstract
:1. Introduction
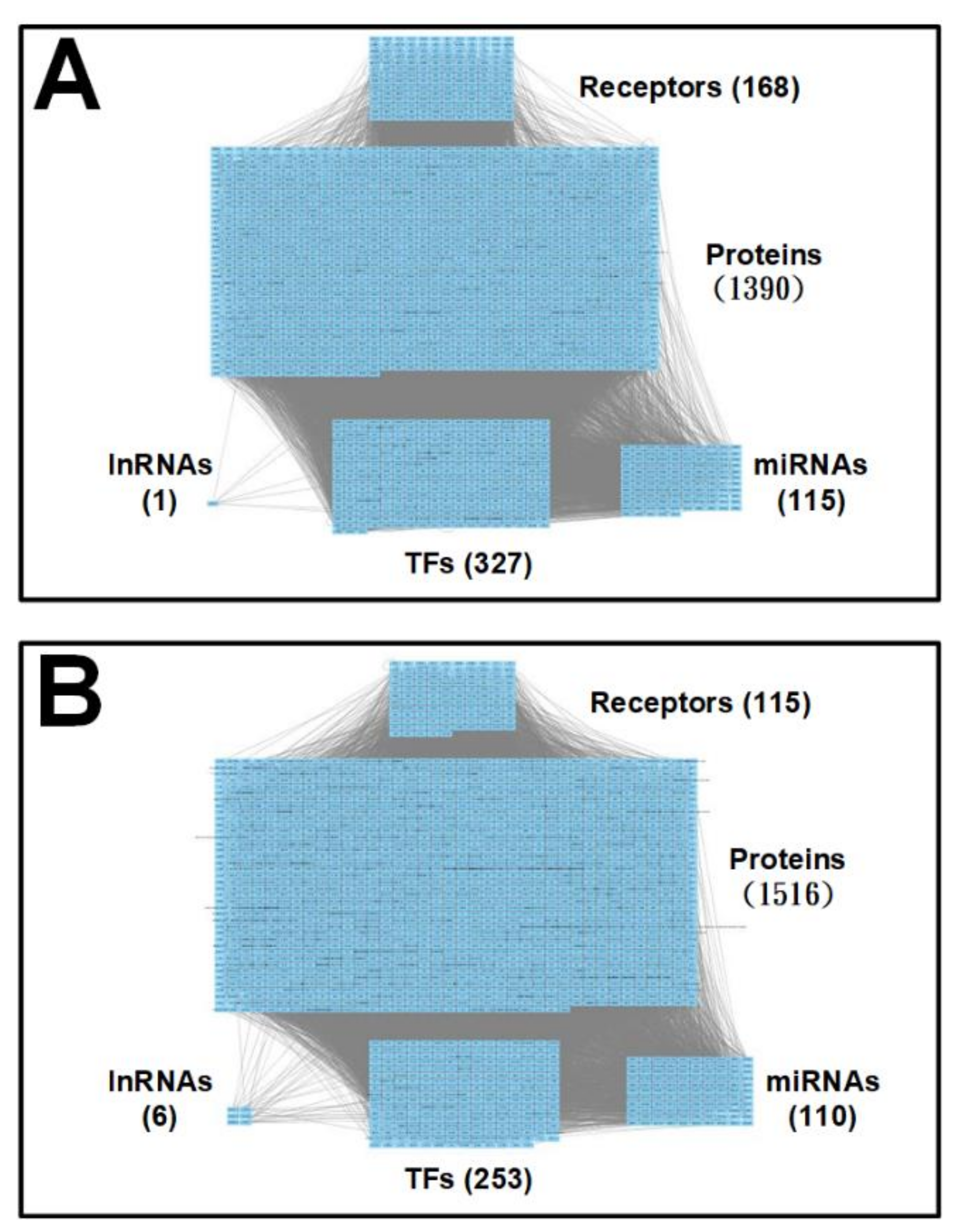
2. Results
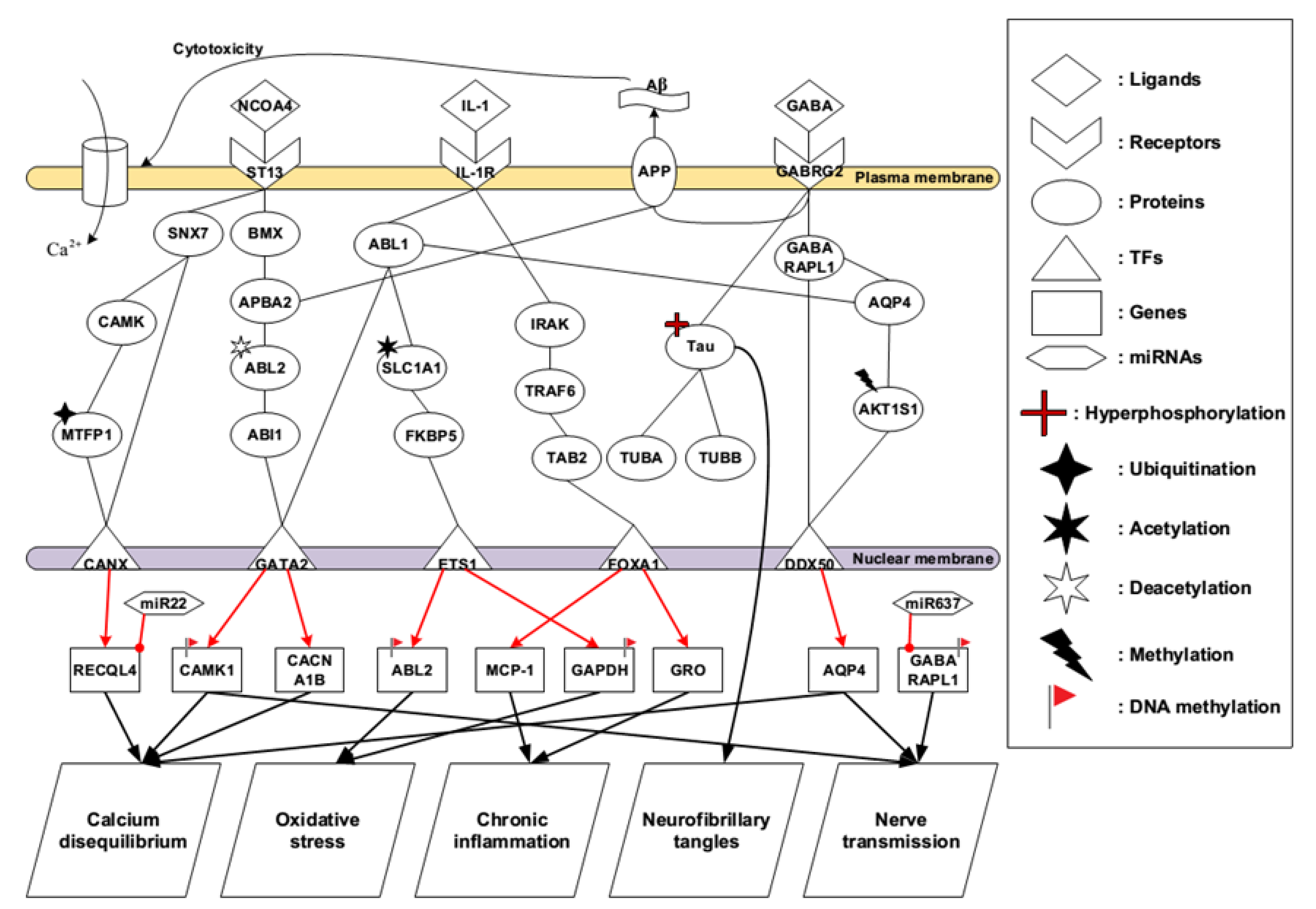
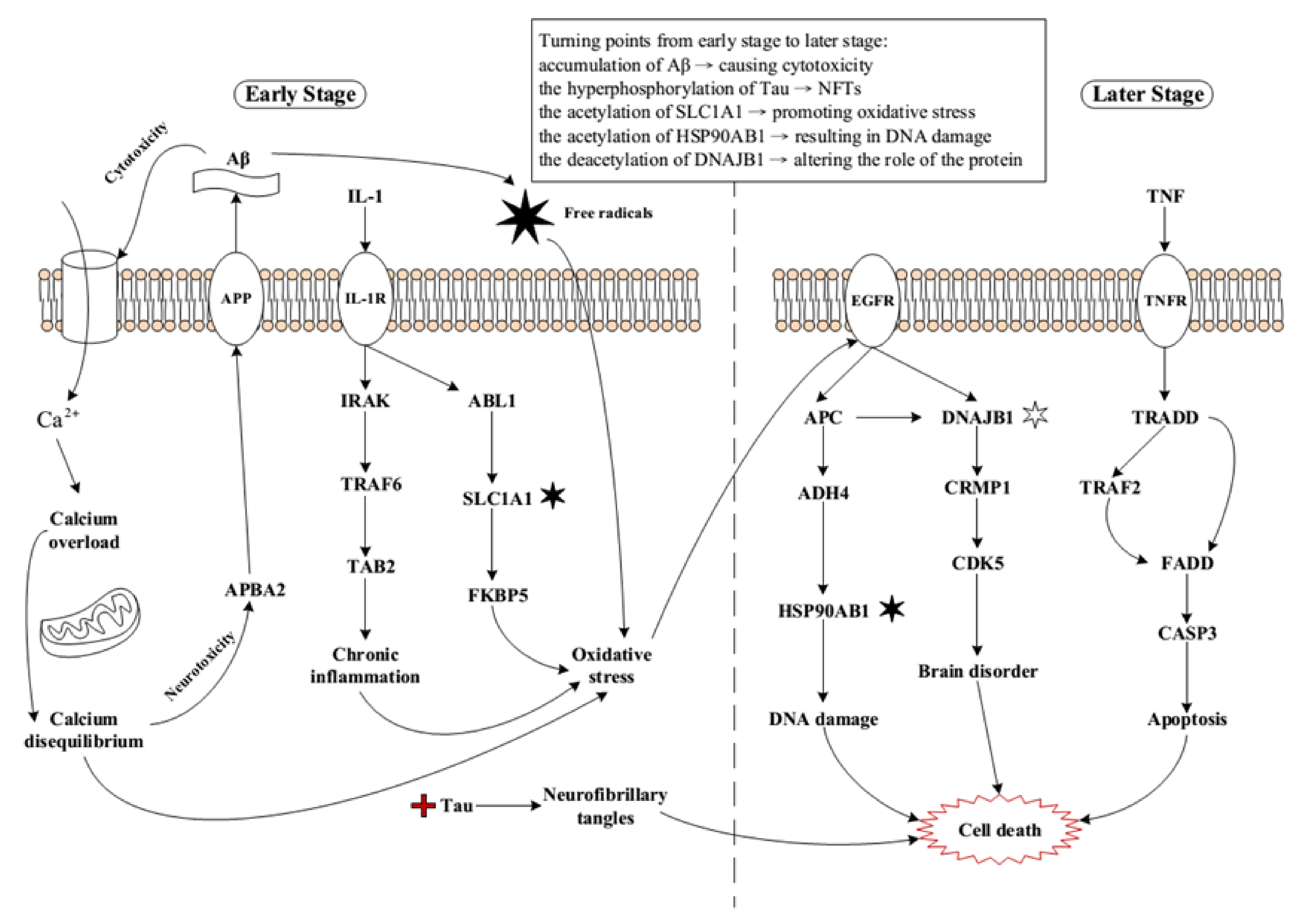
2.1. Core Pathways in Early Stage AD
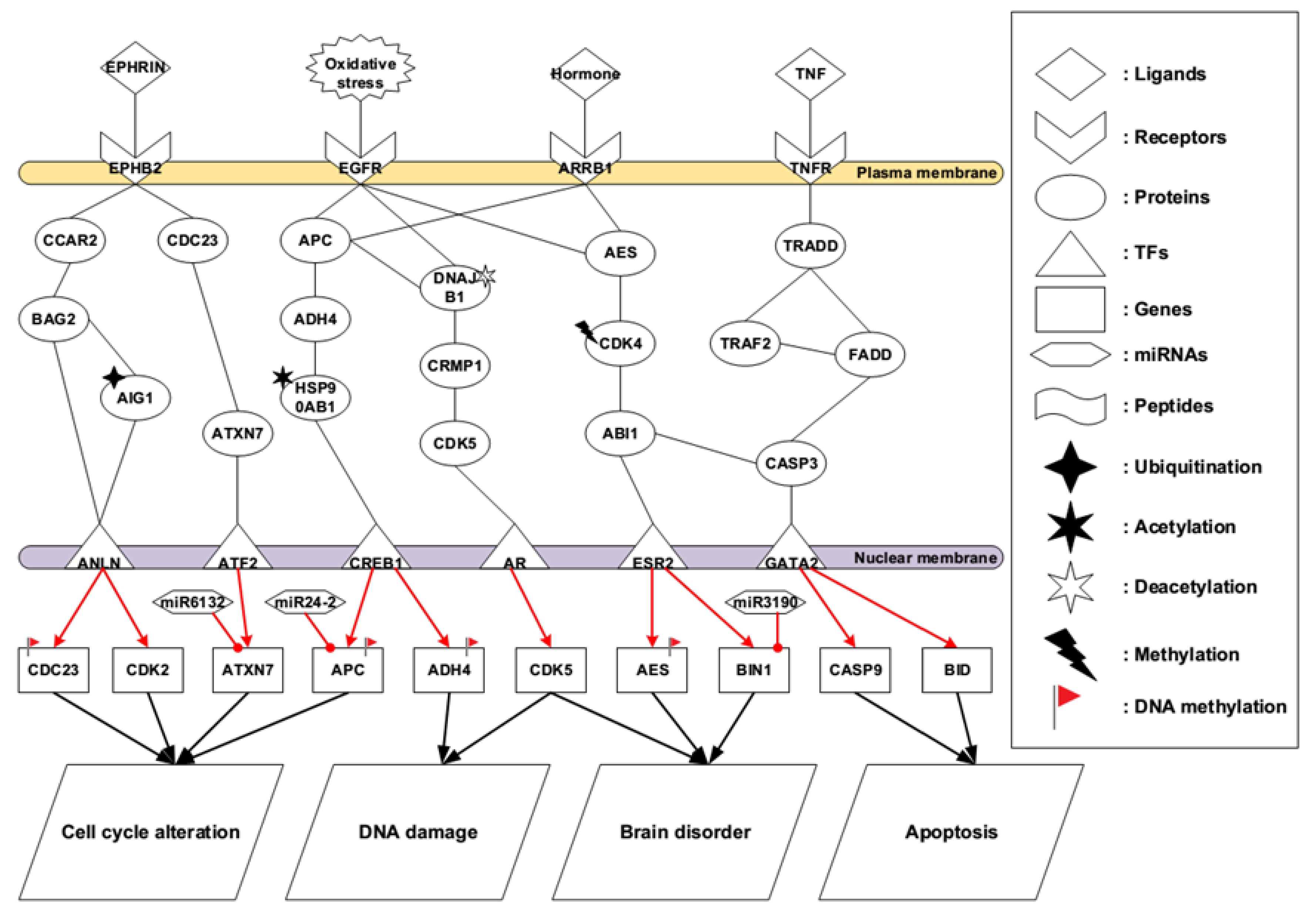
2.2. Core Pathways in Later Stage AD
2.3. Summary of Core Pathways in AD
2.4. Multiple-Molecule Drug Design for the Identified Biomarkers in Early and Later Stage AD
3. Discussion
3.1. Identified Pathogenetic Mechanisms in Early Stage AD
3.2. Identified Pathogenetic Mechanisms in Later Stage AD
4. Materials and Methods
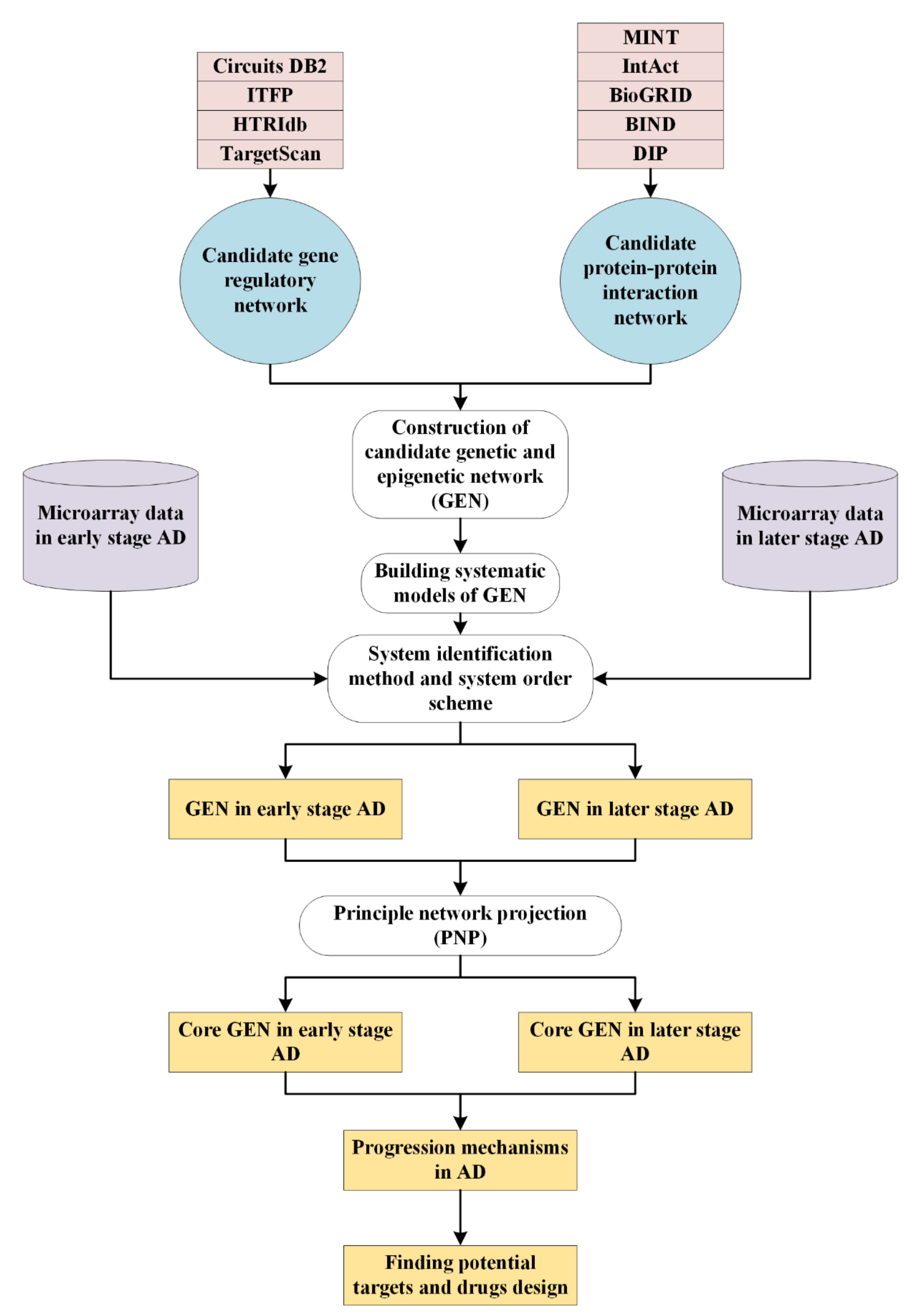
4.1. Overview of Systems Biology Approaches to Investigate Pathogenetic Mechanisms of AD
4.2. Datasets
4.3. Systems Modeling for the Candidate GEN
4.4. Systems Identification to the Candidate GEN via Microarray Data of Early and Later Stage AD
4.5. System Order Detection Scheme for Obtaining Real GEN of Early and Later Stage AD
4.6. PNP Method to Extract the Core GEN from Real GEN for Early and Later Stage AD
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Alzheimer’s disease: Treatments in discovery and development. Nat. Neurosci. 2002, 5, 1055–1057. [Google Scholar] [CrossRef]
- Joachim, C.L.; Selkoe, D.J. The seminal role of! b-amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1992, 6, 7–34. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Schoenberg, B.S.; Kokmen, E.; Okazaki, H. Alzheimer’s disease and other dementing illnesses in a defined United States population: Incidence rates and clinical features. Ann. Neurol. 1987, 22, 724–729. [Google Scholar] [CrossRef]
- Larson, E.B.; Shadlen, M.-F.; Wang, L.; McCormick, W.C.; Bowen, J.D.; Teri, L.; Kukull, W.A. Survival after initial diagnosis of Alzheimer disease. Ann. Intern. Med. 2004, 140, 501–509. [Google Scholar] [CrossRef]
- Walsh, J.S.; Welch, H.G.; Larson, E.B. Survival of outpatients with Alzheimer-type dementia. Ann. Intern. Med. 1990, 113, 429–434. [Google Scholar] [CrossRef]
- Burns, A.; Lewis, G.; Jacoby, R.; Levy, R. Factors affecting survival in Alzheimer’s disease. Psychol. Med. 1991, 21, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Heyman, A.; Peterson, B.; Fillenbaum, G.; Pieper, C. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part XIV: Demographic and clinical predictors of survival in patients with Alzheimer’s disease. Neurology 1996, 46, 656–660. [Google Scholar] [CrossRef]
- Moritz, D.J.; Fox, P.J.; Luscombe, F.A.; Kraemer, H.C. Neurological and psychiatric predictors of mortality in patients with Alzheimer disease in California. Arch. Neurol. 1997, 54, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Lieberburg, I. Cellular mechanisms of β-amyloid production and secretion. Proc. Natl. Acad. Sci. USA 1999, 96, 11049–11053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C. Natural compounds that modulate BACE1-processing of amyloid-beta precursor protein in Alzheimer’s disease. Discov. Med. 2012, 14, 189–197. [Google Scholar]
- Ghosh, A.K.; Brindisi, M.; Tang, J.J.J.o.n. Developing β-secretase inhibitors for treatment of Alzheimer’s disease. J. Neurochem. 2012, 120, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Steele, T.G.; Hills, I.D.; Nomland, A.A.; de León, P.; Allison, T.; McGaughey, G.; Colussi, D.; Tugusheva, K.; Haugabook, S.J.; Espeseth, A.S.J.B.; et al. Identification of a small molecule β-secretase inhibitor that binds without catalytic aspartate engagement. Bioorganic Med. Chem. Lett. 2009, 19, 17–20. [Google Scholar] [CrossRef]
- Forlenza, O.V. Tratamento farmacológico da doença de Alzheimer. Arch. Clin. Psychiatry 2005, 32, 137–148. [Google Scholar] [CrossRef]
- Bonda, D.J.; Wang, X.; Perry, G.; Nunomura, A.; Tabaton, M.; Zhu, X.; Smith, M.A. Oxidative stress in Alzheimer disease: A possibility for prevention. Neuropharmacology 2010, 59, 290–294. [Google Scholar] [CrossRef]
- Richardson, C.; Gard, P.R.; Klugman, A.; Isaac, M.; Tabet, N.J. Blood pro-inflammatory cytokines in Alzheimer’s disease in relation to the use of acetylcholinesterase inhibitors. Int. J. Geriatr. Psychiatry 2013, 28, 1312–1317. [Google Scholar] [CrossRef]
- Mount, C.; Downton, C. Alzheimer disease: Progress or profit? Nat. Med. 2006, 12, 780–784. [Google Scholar] [CrossRef] [Green Version]
- Gordon, M.L.; Kingsley, P.B.; Goldberg, T.E.; Koppel, J.; Christen, E.; Keehlisen, L.; Kohn, N.; Davies, P. An open-label exploratory study with memantine: Correlation between proton magnetic resonance spectroscopy and cognition in patients with mild to moderate Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. Extra 2012, 2, 312–320. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [Green Version]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zawia, N.H.; Lahiri, D.K.; Cardozo-Pelaez, F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic. Biol. Med. 2009, 46, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 2012, 43, 655–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouliaras, L.; Rutten, B.P.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.; van den Hove, D.L. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.-P.; Wang, Y.; Zhang, X.-S.; Xia, W.; Chen, L. Detecting and analyzing differentially activated pathways in brain regions of Alzheimer’s disease patients. Mol. BioSystems 2011, 7, 1441–1452. [Google Scholar] [CrossRef]
- Li, X.; Long, J.; He, T.; Belshaw, R.; Scott, J. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Nikiforova, M.N.; Nikiforov, Y.E. Molecular diagnostics and predictors in thyroid cancer. Thyroid Off. J. Am. Thyroid Assoc. 2009, 19, 1351–1361. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Xing, Y.; Liu, D.; Zhang, R.; Joachimiak, A.; Songyang, Z.; Xu, W. Structural basis of membrane targeting by the Phox homology domain of cytokine-independent survival kinase (CISK-PX). J. Biol. Chem. 2004, 279, 30662–30669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, D.; Guo, F.; Zhu, S.; Xu, X.; Mao, X.; Cao, Y.; Lv, X.; Gao, Q.; Wang, L.; Chen, T.; et al. The alterations of Ca2+/calmodulin/CaMKII/CaV1.2 signaling in experimental models of Alzheimer’s disease and vascular dementia. Neurosci. Lett. 2013, 538, 60–65. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Guo, Y.; Xiao, S. Expression of tyrosine kinase Etk/Bmx and its relationship with AP-1- and NF-kappaB-associated proteins in hepatocellular carcinoma. Oncology 2007, 72, 410–416. [Google Scholar] [CrossRef]
- Miller, C.C.; McLoughlin, D.M.; Lau, K.F.; Tennant, M.E.; Rogelj, B. The X11 proteins, Abeta production and Alzheimer’s disease. Trends Neurosci. 2006, 29, 280–285. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Ann. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Oklinski, M.K.; Skowronski, M.T.; Skowronska, A.; Rützler, M.; Nørgaard, K.; Nieland, J.D.; Kwon, T.H.; Nielsen, S. Aquaporins in the Spinal Cord. Int. J. Mol. Sci. 2016, 17, 2050. [Google Scholar] [CrossRef] [Green Version]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Sun, X.; Jin, L.; Ling, P. Review of drugs for Alzheimer’s disease. Drug Discov. Ther. 2012, 6, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Supnet, C.; Bezprozvanny, I. Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2010, 20 (Suppl. S2), S487–S498. [Google Scholar] [CrossRef] [Green Version]
- Itkin, A.; Dupres, V.; Dufrêne, Y.F.; Bechinger, B.; Ruysschaert, J.M.; Raussens, V. Calcium ions promote formation of amyloid β-peptide (1–40) oligomers causally implicated in neuronal toxicity of Alzheimer’s disease. PLoS ONE 2011, 6, e18250. [Google Scholar] [CrossRef] [Green Version]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Mudher, A.; Lovestone, S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 2002, 25, 22–26. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Walker, D.; McGeer, E.; McGeer, P. Involvement of inflammation and complement in Alzheimer’s disease. Clin. Neuroimmunol. 1997, 172–188. [Google Scholar]
- Jovicic, A.; Zaldivar Jolissaint, J.F.; Moser, R.; Silva Santos Mde, F.; Luthi-Carter, R. MicroRNA-22 (miR-22) overexpression is neuroprotective via general anti-apoptotic effects and may also target specific Huntington’s disease-related mechanisms. PLoS ONE 2013, 8, e54222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, W.H.; Kolar, M.J.; Kamat, S.S.; Cognetta 3rd, A.B.; Hulce, J.J.; Saez, E.; Kahn, B.B.; Saghatelian, A.; Cravatt, B.F. AIG1 and ADTRP are atypical integral membrane hydrolases that degrade bioactive FAHFAs. Nat. Chem. Biol. 2016, 12, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Sikorski, R.S.; Boguski, M.S.; Goebl, M.; Hieter, P. A repeating amino acid motif in CDC23 defines a family of proteins and a new relationship among genes required for mitosis and RNA synthesis. Cell 1990, 60, 307–317. [Google Scholar] [CrossRef]
- Meng, J.; Liu, Y.; Han, J.; Tan, Q.; Chen, S.; Qiao, K.; Zhou, H.; Sun, T.; Yang, C. Hsp90β promoted endothelial cell-dependent tumor angiogenesis in hepatocellular carcinoma. Mol. Cancer 2017, 16, 72. [Google Scholar] [CrossRef] [Green Version]
- Dubielecka, P.M.; Cui, P.; Xiong, X.; Hossain, S.; Heck, S.; Angelov, L.; Kotula, L. Differential regulation of macropinocytosis by Abi1/Hssh3bp1 isoforms. PLoS ONE 2010, 5, e10430. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Gaur, U.; Aggarwal, B.B. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem. Pharmacol. 2003, 66, 1403–1408. [Google Scholar] [CrossRef]
- Sana, J.; Hajduch, M.; Michalek, J.; Vyzula, R.; Slaby, O. MicroRNAs and glioblastoma: Roles in core signalling pathways and potential clinical implications. J. Cell. Mol. Med. 2011, 15, 1636–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sourbier, C.; Ricketts, C.J.; Matsumoto, S.; Crooks, D.R.; Liao, P.J.; Mannes, P.Z.; Yang, Y.; Wei, M.H.; Srivastava, G.; Ghosh, S.; et al. Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer Cell 2014, 26, 840–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.G.; Ryan, R.M.; Thoeng, A.D.; Ng, C.; King, K.; Vanslambrouck, J.M.; Auray-Blais, C.; Vandenberg, R.J.; Bröer, S.; Rasko, J.E. Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J. Clin. Investig. 2011, 121, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groden, J.; Thliveris, A.; Samowitz, W.; Carlson, M.; Gelbert, L.; Albertsen, H.; Joslyn, G.; Stevens, J.; Spirio, L.; Robertson, M.; et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66, 589–600. [Google Scholar] [CrossRef]
- Neniskyte, U.; Vilalta, A.; Brown, G.C. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014, 588, 2952–2956. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.A.; Alleva, L.M.; Vissel, B. The roles of TNF in brain dysfunction and disease. Pharmacol. Ther. 2010, 128, 519–548. [Google Scholar] [CrossRef]
- Cotman, C.W.; Su, J.H. Mechanisms of neuronal death in Alzheimer’s disease. Brain Pathol. 1996, 6, 493–506. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.; Subramanian, A. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Musa, A.; Ghoraie, L.S.; Zhang, S.D.; Glazko, G.; Yli-Harja, O.; Dehmer, M.; Haibe-Kains, B.; Emmert-Streib, F. A review of connectivity map and computational approaches in pharmacogenomics. Brief. Bioinform. 2018, 19, 506–523. [Google Scholar] [CrossRef] [Green Version]
- Detke, M.J.; Lu, Y.; Goldstein, D.J.; Hayes, J.R.; Demitrack, M.A. Duloxetine, 60 mg once daily, for major depressive disorder: A randomized double-blind placebo-controlled trial. J. Clin. Psychiatry 2002, 63, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.B.; Rasmusson, D.X.; Folstein, M.F.; Carson, K.A.; Kawas, C.; Brandt, J. Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology 1995, 45, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, W.B. A review of the evidence for the efficacy and safety of trazodone in insomnia. J. Clin. Psychiatry 2005, 66, 469–476. [Google Scholar] [CrossRef] [PubMed]
- del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andrés, M.V.; Gómez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; León, T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J. Alzheimer’s Dis. JAD 2013, 33, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Carroll, J.; Trojanowski, J.Q.; Yao, Y.; Iba, M.; Potuzak, J.S.; Hogan, A.M.; Xie, S.X.; Ballatore, C.; Smith 3rd, A.B.; et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef]
- Mohs, R.; Greig, N. Drug discovery and development: Role of basic biological research. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 651–657. [Google Scholar] [CrossRef]
- Coric, V.; Salloway, S.; van Dyck, C.H.; Dubois, B.; Andreasen, N.; Brody, M.; Curtis, C.; Soininen, H.; Thein, S.; Shiovitz, T.; et al. Targeting Prodromal Alzheimer Disease with Avagacestat: A Randomized Clinical Trial. JAMA Neurol. 2015, 72, 1324–1333. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.-J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A Phase II Trial of Tideglusib in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Zempel, H.; Mandelkow, E.M. Tau missorting and spastin-induced microtubule disruption in neurodegeneration: Alzheimer Disease and Hereditary Spastic Paraplegia. Mol. Neurodegener. 2015, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.A.; Chuckowree, J.A.; Dyer, M.S.; Dickson, T.C.; Blizzard, C.A. Epothilone D alters normal growth, viability and microtubule dependent intracellular functions of cortical neurons in vitro. Sci. Rep. 2020, 10, 918. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef]
- Nixon, R.A. The calpains in aging and aging-related diseases. Ageing Res. Rev. 2003, 2, 407–418. [Google Scholar] [CrossRef]
- McKee, A.C.; Kosik, K.S.; Kennedy, M.B.; Kowall, N.W. Hippocampal neurons predisposed to neurofibrillary tangle formation are enriched in type II calcium/calmodulin-dependent protein kinase. J. Neuropathol. Exp. Neurol. 1990, 49, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Migliore, L.; Fontana, I.; Trippi, F.; Colognato, R.; Coppedè, F.; Tognoni, G.; Nucciarone, B.; Siciliano, G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol. Aging 2005, 26, 567–573. [Google Scholar] [CrossRef]
- Fetler, L.; Amigorena, S. Neuroscience. Brain under surveillance: The microglia patrol. Science 2005, 309, 392–393. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.J.; Drego, R.; Hakimian, E.; Masliah, E. Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr. Drug Targets Inflamm. Allergy 2005, 4, 247–256. [Google Scholar] [CrossRef]
- Lanzrein, A.S.; Johnston, C.M.; Perry, V.H.; Jobst, K.A.; King, E.M.; Smith, A.D. Longitudinal study of inflammatory factors in serum, cerebrospinal fluid, and brain tissue in Alzheimer disease: Interleukin-1beta, interleukin-6, interleukin-1 receptor antagonist, tumor necrosis factor-alpha, the soluble tumor necrosis factor receptors I and II, and alpha1-antichymotrypsin. Alzheimer Dis. Assoc. Disord. 1998, 12, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Mammalian G1 cyclins and cell cycle progression. Proc. Assoc. Am. Physicians 1995, 107, 181–186. [Google Scholar] [PubMed]
- Giovanni, A.; Wirtz-Brugger, F.; Keramaris, E.; Slack, R.; Park, D.S. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J. Biol. Chem. 1999, 274, 19011–19016. [Google Scholar] [CrossRef] [Green Version]
- Lauterbach, E.C.; Victoroff, J.; Coburn, K.L.; Shillcutt, S.D.; Doonan, S.M.; Mendez, M.F. Psychopharmacological neuroprotection in neurodegenerative disease: Assessing the preclinical data. J. Neuropsychiatry Clin. Neurosci. 2010, 22, 8–18. [Google Scholar] [CrossRef]
- El-Guendy, N.; Rangnekar, V.M. Apoptosis by Par-4 in cancer and neurodegenerative diseases. Exp. Cell Res. 2003, 283, 51–66. [Google Scholar] [CrossRef]
- Xenarios, I.; Rice, D.W.; Salwinski, L.; Baron, M.K.; Marcotte, E.M.; Eisenberg, D. DIP: The database of interacting proteins. Nucleic Acids Res. 2000, 28, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Bader, G.D.; Betel, D.; Hogue, C.W. BIND: The Biomolecular Interaction Network Database. Nucleic Acids Res. 2003, 31, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Chatr-aryamontri, A.; Breitkreutz, B.J.; Chang, C.S.; Rust, J.M.; Theesfeld, C.L.; Heinicke, S.; Breitkreutz, A.; Chen, D.; Hirschman, J.; et al. BioGRID: A Resource for Studying Biological Interactions in Yeast. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Kerrien, S.; Alam-Faruque, Y.; Aranda, B.; Bancarz, I.; Bridge, A.; Derow, C.; Dimmer, E.; Feuermann, M.; Friedrichsen, A.; Huntley, R.; et al. IntAct—Open source resource for molecular interaction data. Nucleic Acids Res. 2006, 35, D561–D565. [Google Scholar] [CrossRef]
- Zanzoni, A.; Montecchi-Palazzi, L.; Quondam, M.; Ausiello, G.; Helmer-Citterich, M.; Cesareni, G. MINT: A Molecular INTeraction database. FEBS Lett. 2002, 513, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Friard, O.; Re, A.; Taverna, D.; De Bortoli, M.; Corá, D. Circuits DB: A database of mixed microRNA/transcription factor feed-forward regulatory circuits in human and mouse. BMC Bioinform. 2010, 11, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Tu, K.; Yang, Q.; Xiong, Y.; Wei, C.; Xie, L.; Zhu, Y.; Li, Y. ITFP: An integrated platform of mammalian transcription factors. Bioinformatics 2008, 24, 2416–2417. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E.; Dietze, P.; Karas, H.; Knüppel, R. TRANSFAC: A Database on Transcription Factors and Their DNA Binding Sites. Nucleic Acids Res. 1996, 24, 238–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bovolenta, L.A.; Acencio, M.L.; Lemke, N. HTRIdb: An open-access database for experimentally verified human transcriptional regulation interactions. BMC Genom. 2012, 13, 405. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Ishiguro, M.; Kitagawa, G.J.D. Akaike Information Criterion Statistics; D. Reidel: Dordrecht, The Netherlands, 1986; Volume 81, p. 26853. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Description | |
|---|---|---|
| Duloxetime | The number of targets | 8 |
| Targets to be inhibited | ST13, IL-1R, CAMK1, SLC1A1, AQP4, TAB2 | |
| Targets to be enhanced | GABRG2, APBA2 | |
| Sulindac | The number of targets | 7 |
| Targets to be inhibited | ST13, IL-1R, CAMK1, SLC1A1, AQP4 | |
| Targets to be enhanced | GABRG2, APBA2 | |
| Avagacestat | The number of targets | 6 |
| Targets to be inhibited | ST13, IL-1R, CAMK1, SLC1A1, TAB2 | |
| Targets to be enhanced | GABRG2 | |
| Drug | Description | |
|---|---|---|
| Trazodone | The number of targets | 8 |
| Targets to be inhibited | EPHB2, EGFR, ARRB1, TNFR, FADD | |
| Targets to be enhanced | CDK5, AIG1, APC | |
| Tideglusib | The number of targets | 7 |
| Targets to be inhibited | EPHB2, EGFR, ARRB1, TNFR, FADD | |
| Targets to be enhanced | CDK5, APC | |
| Epothilone D | The number of targets | 7 |
| Targets to be inhibited | EPHB2, EGFR, ARRB1, TNFR, FADD | |
| Targets to be enhanced | CDK5, AIG1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, S.-J.; Chung, M.-H.; Chen, B.-S. Investigating Pathogenetic Mechanisms of Alzheimer’s Disease by Systems Biology Approaches for Drug Discovery. Int. J. Mol. Sci. 2021, 22, 11280. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011280
Yeh S-J, Chung M-H, Chen B-S. Investigating Pathogenetic Mechanisms of Alzheimer’s Disease by Systems Biology Approaches for Drug Discovery. International Journal of Molecular Sciences. 2021; 22(20):11280. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011280
Chicago/Turabian StyleYeh, Shan-Ju, Ming-Hsun Chung, and Bor-Sen Chen. 2021. "Investigating Pathogenetic Mechanisms of Alzheimer’s Disease by Systems Biology Approaches for Drug Discovery" International Journal of Molecular Sciences 22, no. 20: 11280. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222011280